Что такое буллёзный эпидермолиз? | Университетская клиника г. Фрайбурга
Уход за новорожденными с подозрением на буллезный эпидермолиз
Основные правила
Максимально осторожное и нежное обращение с новорожденными и младенцами. Необходимо свести к минимуму любые травмирующие кожу факторы, т. к. это может спровоцировать возникновение пузырей. Нельзя использовать обычные перевязочные материалы. Скрининг новорожденного должен проводится путем осторожного забора крови из вены. При подозрении на наличие буллезного эпидермолиза следует как можно скорее провести диагностические исследования, включающие в себя биопсию кожи и анализ на мутации.
Общие рекомендации
Не использовать обычный пластырь или другие обычные самоклеящиеся повязки. Если они все же по ошибке были использованы, их следует очень аккуратно удалить с помощью специальных растворяющих средств (напр., Niltac).
При мониторинге или проведении ЭКГ не наклеивать электроды непосредственно на кожу, а осуществлять наблюдение с помощью пульсоксиметрии или фиксировать электроды с помощью силиконовых лент Mepilex transfer или Mepitac.
После купания не растирать кожу ребенка, а очень аккуратно обсушивать.
Ни в коем случае не поднимать и не носить ребенка, взяв под мышки. Чтобы поднять ребенка, следует сначала подложить под его тело пеленку, затем повернуть его на бок, подложить под пеленку руки таким образом, чтобы одна рука поддерживала голову и плечи, а вторая – нижнюю часть туловища ребенка. Поднимать ребенка как можно осторожнее, одним движением. Укладывать ребенка рекомендуется на ватный или мягкий матрас из пеноматериала, во время пребывания в клинике использовать специальный противопролежневый матрас из вискоэластика. При транспортировке следует выложить внутреннюю поверхность переносной колыбели мягким пеноматериалом (напр. Ligasano), чтобы предотвратить образование пузырей при усиленном потоотделении.
Одежда ребенка должна быть свободной и мягкой, все предметы одежды должны одеваться швами наружу, предпочтение следует отдавать ползункам с интегрированным носком.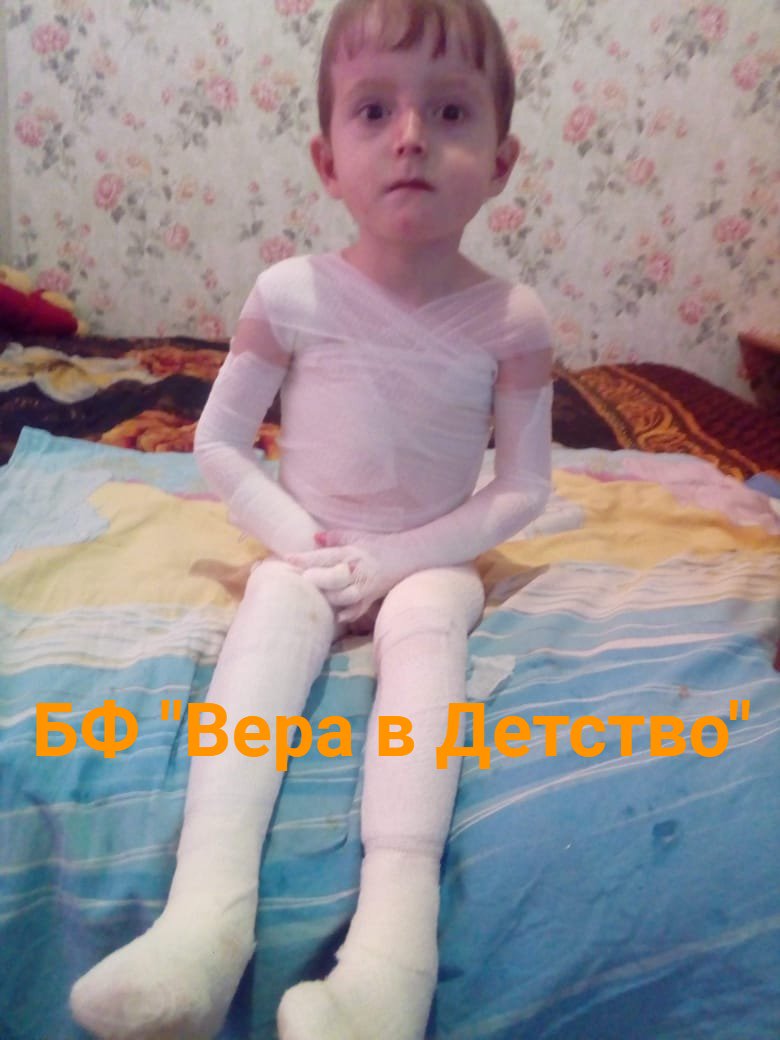 Не одевать ребенка слишком тепло, это может вызвать усиленное потение и спровоцировать образование пузырей. Носочки должны быть без тугих резинок и не давить на кожу. Имеющиеся эластичные резинки следует отрезать. Обувь ребенка должна быть свободной и мягкой.
Не одевать ребенка слишком тепло, это может вызвать усиленное потение и спровоцировать образование пузырей. Носочки должны быть без тугих резинок и не давить на кожу. Имеющиеся эластичные резинки следует отрезать. Обувь ребенка должна быть свободной и мягкой.
Кормление/питание
Для кормления и для успокоения ребенка можно использовать соски и пустышки Habermann. Если ребенок в состоянии брать грудь, возможно также и грудное вскармливание. В таком случае вокруг рта и на щеки ребенка, а также вокруг соска матери необходимо нанести вазелин или мазь бепантен, что позволяет минимизировать трение кожи.
Если ребенок не голоден, однако ему требуется успокоительное сосание, можно предложить ему успокоительную соску-пустышку. Для этого рекомендуется использовать соски из натурального латекса с мягкой эластичной защитной пластинкой. Тем не менее, надо иметь в виду, что сосание пустышки также может способствовать образованию пузырей. Начиная с 10-го дня жизни требуется проводить заместительную терапию витамином Д, а также препаратами фтора. Прием фтора необходим в связи с тем, что гигиена полости рта у пациентов с БЭ может быть сильно затруднена.
Требуется особенно тщательно наблюдать за ростом и развитием ребенка с заболеванием БЭ, т.к. постоянные процессы заживления кожи требуют повышенного расхода калорий. Детей нужно регулярно взвешивать. При недостаточном развитии ребенка по согласованию с педиатром необходимо дополнительно к материнскому молоку ввести в его рацион специальные питательные добавки (напр. Maltodextrin).
Частым осложнением БЭ являются запоры. Если достаточное количество жидкости и специальное питание не способствуют смягчению консистенции стула, то назначаются не свечи, а оральные препараты (напр. Lactulose).
Общие правила ухода за кожей, пупком и руками
В целом, необходимости в специальном уходе за здоровыми участками кожи младенцев нет. Важно обрабатывать корочки, образующиеся в процессе заживления пузырей, смягчающими мазями (напр. Linola, Bepanthen) и затем аккуратно удалять их, т.к. в противном случае они могут вызывать раздражение кожи и возникновенение новых пузырьков.
Linola, Bepanthen) и затем аккуратно удалять их, т.к. в противном случае они могут вызывать раздражение кожи и возникновенение новых пузырьков.
До отпадения пуповины с целью предотвращения трения и возникновения пузырей необходимо непосредственно вокруг пупка накладывать силиконовую губчатую повязку (напр. Mepilex lite или transfer).
Ногти младенцам следует обрезать только тогда, когда они станут очень длинными и появится опасность, что ребенок сможет поранить себя. Следует учитывать то, что сам процесс обрезания также может вызвать образование пузырей, поэтому по возможности делать это следует во время сна ребенка.
Если ребенок сосет пальцы, то на ручки надо одевать специальные варежки или носочки.
Обработка пузырей и ран
Пузыри следует прокалывать большой стерильной канюлей в нескольких местах, за 2 минуты до этого детям грудного возраста с целью обезболивания следует дать 1 – 2 мл G40% орально. При уходе за эрозиями и пузырями нельзя использовать сухие повязки, т.к. они могут приклеиваться и в дальнейшем при замене повреждать кожу и провоцировать возникновение новых пузырей.
При инфицированных, гнойных пузырях, покрывающую их кожу необходимо удалить и продезинфицировать рану.
На свежие раны рекомендуется накладывать сетчатую накладку Urgotül или Mepitel, затем внешнюю повязку из силиконового губчатого материала (Mepilex lite, Mepilex transfer) и далее фиксировать все мягкой повязкой (напр. Medicomp), марлевым бинтом или трубчатой фиксирующей повязкой. Если имеется необходимость наклеить пластырь непосредственно на кожу, можно использовать повязку Mepitac.
При неинфицированных ранах, чтобы максимально оградить ребенка от болей, вызываемых процессом перевязки, мепитекс можно оставить на несколько дней (макс. 7 дней). Внешнюю повязку требуется менять каждый день.
При сухих открытых ранах для обеспечения увлажнения на или под сетчатую накладку можно наносить мази Bepanten или Prontosan.
Для минимизации травмирования кожи и предотвращения образования пузырей в местах преимущественной локализации (в области ягодиц, пяток и резинок подгузников) рекомендуется использовать повязки Mepilex, Mepilex lite и transfer.
При подозрении на инфицирование раны при каждой смене повязки ее следует в течение 5 минут обработать дезинфицирующим раствором (напр. Octenisept, Prontosan, Lavanid). Если это не приведет к улучшению состояния раны или появится жар, необходимо взять мазок и провести оральную антибиотическую терапию (напр. препаратом Cefalosporin 2-го поколения).
От повязок с содержанием серебра из-за опасности возникновения аргирии в первые месяцы жизни ребенка следует отказаться.
Если новорожденный содержится в инкубаторе, необходимо обязательно следить за тем, чтобы в нем не застаивался слишком теплый воздух, т.к. это увеличивает риск образования пузырей.
Обработка пузырей в области подгузников
Необходимо стараться максимально избегать трения в области подгузников, а также сильно не растирать кожу при ее обработке и очищении. Труднодоступные для очищения участки необходимо покрыть марлевой повязкой, пропитанной слоем Bepanten, и затем использовать ее для очищения при смене подгузников.
Если у детей в области подгузников образовались пузыри, необходимо накладывать силиконовые повязки (Mepilex transfer). Эластичные резинки от подгузников рекомендуется срезать.
Перианальные трещины смазывать цинковой пастой. В остальном, применяются вышеуказанные общие правила обработки ран и ухода за кожей.
причины, симптомы, лечение, профилактика — клиника «Добробут»
Буллезный эпидермолиз: причины, симптомы, лечение
Буллезный эпидермолиз – это не одна патология, а целая группа наследственных нозологий, которые манифестируются легкой ранимостью кожных покровов. Основное проявление – формирование на коже пузырей с жидким содержимым, а после их вскрытия – образование эрозий, которые долго не заживают.
Причины и формы буллезного эпидермолиза
Причины буллезного эпидермолиза зависят от типа.
Простой буллезный эпидермолиз развивается из-за мутаций генов KRT5 и KRT14.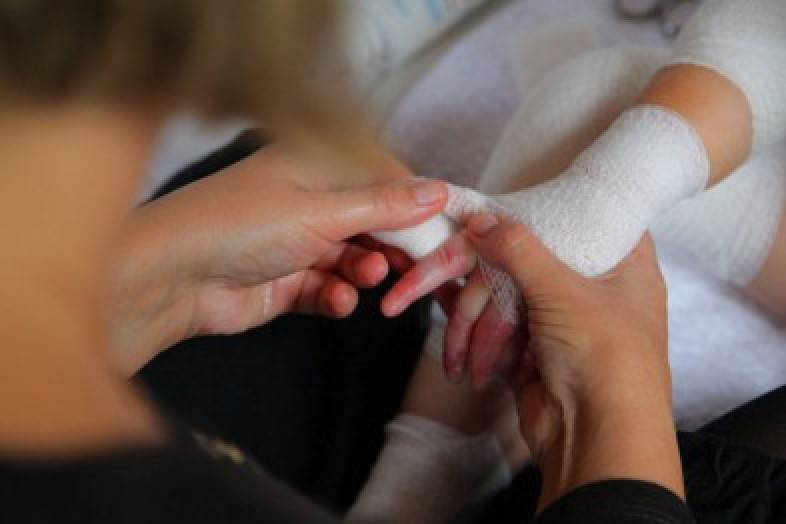 Предполагается, что при этом в кожных покровах нарушается равновесие между ферментами и ингибиторами (соединениями, способными подавлять процессы в тканях). Как результат, выделяются ферменты, которые разрушают белки кожи, на фоне чего образуются пузыри.
Предполагается, что при этом в кожных покровах нарушается равновесие между ферментами и ингибиторами (соединениями, способными подавлять процессы в тканях). Как результат, выделяются ферменты, которые разрушают белки кожи, на фоне чего образуются пузыри.
Пограничный буллезный эпидермолиз провоцируют мутации в генах под маркировкой LAMB3 и LAMA3. Курируемая ими ферментная система становится разбалансированной, из-за чего страдают коллаген 17-го типа и ламинин-332, без которых нарушается нормальное строение кожи. Помимо формирования пузырей и эрозий, появляется усиленная ломкость кожи.
Дистрофический буллезный эпидермолиз развивается из-за мутаций в гене COL7A1. Из-за этого страдает коллаген 7-го типа, контролирующий состояние соединительнотканных элементов кожи. Нехватка этого протеина провоцирует образование на коже сыпи, эрозий (язвочек) и пузырей.
Особенностью смешанного буллезного эпидермолиза является формирование пузырей во всех слоях кожи.
Это основные типы патологии. На данный момент различают десятки видов буллезного эпидермолиза.
Симптомы
Симптомы патологии могут отличаться, но общий признак разных форм буллезного эпидермолиза – образование пузырей и язвочек при механическом воздействии на кожу.
При локализованной форме простого буллезного эпидермолиза изменения кожи наблюдаются только на одном участке тела (кисти, стопе).
Пограничная форма буллезного эпидермолиза характеризуется более тяжелым состоянием. Например, при летальном подтипе Херлитца диагностируются:
- усиленная ломкость кожи;
- формирование большого количества пузырей и эрозий;
- образование грануляций на лице и спине.
Нередко больные буллезным эпидермолизом летального подтипа умирают в первые годы жизни. У выживших наблюдаются:
- контрактуры (тугоподвижность) суставов;
- поражение почек;
- потеря ногтевых пластин.
Для атрофической формы пограничного буллезного эпидермолиза характерны обширные высыпания с образованием рубцов.
Дистрофический буллезный эпидермолиз в основном поражает большие участки тела. Его доминантный вариант (связанный с доминантными генами) более доброкачественный – такие больные теряют ногти, у них образуются заметные рубцы. Рецессивный вариант более тяжелый: при нем поражаются кости, а на месте шрамов с годами может возникнуть плоскоклеточный рак.
Диагностика
Диагноз буллезного эпидермолиза у детей и взрослых ставят на основании осмотра кожи, проведения иммуногистологических исследований и генетического анализа, наследственного анамнеза.
Главный тест заключается в том, что врач механически воздействует на кожу пациента и через некоторое время оценивает последствия такого раздражения.
При иммунофлуоресцентном анализе используют антитела, которые имеют сродство к белкам кожи. При помощи такого метода можно оценить количество белков – а значит, и ферментную активность тканей. Сниженный уровень белка подтверждает его низкое выделение либо форсированное разрушение.
Изучение наследственного анамнеза помогает выявить у пациента родственников с таким же заболеванием.
Осложнения
Важно знать о причинах буллезного эпидермолиза, а также возможных осложнениях. Буллезный эпидермолиз независимо от вида чаще всего сопровождается такими осложнениями:
- присоединение вторичной инфекции;
- инфекционно-токсический шок;
- сепсис;
- обезвоживание.
Буллезный эпидермолиз: лечение
Специфическое лечение не разработано. Цель терапевтических процедур – уменьшение выраженности кожных нарушений и предупреждение формирования осложнений.
При развитии тяжелых форм патологии назначают преднизолон.
Местное лечение заключается во вскрытии пузырьков, их обработке антисептиками. Повязку накладывают очень осторожно – давление способно спровоцировать развитие новых пузырей.
Также разрабатывают методы лечения с применением стволовых клеток, белковую и генную терапии – но пока они апробируются только на животных. На данный момент буллезный эпидермолиз является неизлечимой патологией.
На данный момент буллезный эпидермолиз является неизлечимой патологией.
Профилактика
Заболевание является врожденным, поэтому специфической профилактики нет. Для предупреждения развития проблем с кожей на фоне такой патологии следует очень бережно относиться к собственным кожным покровам – избегать травматизации.
Читайте детальнее о лечении буллезного эпидермолиза на нашем сайте Добробут.ком.
Дистрофический буллезный эпидермолиз в сочетании с врожденными контрактурами верхних и нижних конечностей Текст научной статьи по специальности «Клиническая медицина»
ОБЗОР ЛИТЕРАТУРЫ
УДК 617.56/57-009.12-053.1-02:616.529.1(048.8) DOI: 10.17816/PT0RS3451-59
ДИСТРОФИЧЕСКИЙ БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ В СОЧЕТАНИИ С ВРОЖДЕННЫМИ КОНТРАКТУРАМИ ВЕРХНИХ И НИЖНИХ КОНЕЧНОСТЕЙ
© Агранович О.Е., Буклаев Д.С., Тихоненко Т.И.
ФГБУ «НИДОИ им. Г. И. Турнера» Минздрава России, Санкт-Петербург
Буллезный эпидермолиз (БЭ)— это редкое наследственное заболевание, его главный признак — образование пузырей и мокнущих ран (эрозий) на коже и слизистых оболочках, возникающих при незначительном травмировании. Клинические проявления заболевания могут варьировать от локализованных пузырей на руках и стопах до генерализованных высыпаний по всему кожному покрову, а также с поражением слизистой оболочки внутренних органов. В настоящее время выделено четыре основные группы БЭ: простой, промежуточный, дистрофический и синдром Киндлера. Мутации вызывают изменения в структуре белков, ответственных за адгезию между слоями дермы, что и приводит к образованию везикул. Лечение БЭ представляет собой сложную задачу вследствие отсутствия возможности прямого воздействия на патогенез заболевания, и его основной целью является купирование существующих кожных проявлений и предотвращение появления новых элементов. 4-интегрина или одну из трех цепей ла-минина-332, что вызывает нарушение формирования гемидесмосом и фиксации соединительных волокон. Клинически определяются множественные буллезные элементы, имеющие большую площадь поражения кожи. Характерной особенностью данного типа БЭ является образование грануляционной ткани в области лица, спины, подмышечных впадин. Выделяют три основных подтипа ПоБЭ: Херлитца, не-Херлитца и ПоБЭ с атрезией пилорического отдела желудка [4, 6, 7].
4-интегрина или одну из трех цепей ла-минина-332, что вызывает нарушение формирования гемидесмосом и фиксации соединительных волокон. Клинически определяются множественные буллезные элементы, имеющие большую площадь поражения кожи. Характерной особенностью данного типа БЭ является образование грануляционной ткани в области лица, спины, подмышечных впадин. Выделяют три основных подтипа ПоБЭ: Херлитца, не-Херлитца и ПоБЭ с атрезией пилорического отдела желудка [4, 6, 7].
Подтип Херлитца — наиболее тяжелый генерализованный вариант буллезного эпидермоли-за. Обширные раневые дефекты определяются с рождения ребенка и обусловливают частые септические осложнения, тяжелые белково-электро-литные нарушения, обезвоживание, выраженную гипотрофию. Наличие внекожных буллезных элементов (в области пищевода, желудка, дыхательных путей, кишечника, мочеполовой системы) приводит к тяжелой полиорганной недостаточности, и нередко дети погибают в возрасте до двух лет.
При подтипе не-Херлитца у пациентов, кроме генерализованных пузырей, определяются грубые, утолщенные ногтевые пластинки, атрофические рубцы, нарушение формирования зубной эмали,
рубцовые аллопеции. Однако внекожные проявления за исключением стеноза гортани и трахеи встречаются крайне редко.
Дистрофический буллезный эпидермолиз (ДБЭ, DEB) наследуется как по аутосомно-доми-нантному (ДДБЭ), так и по аутосомно-рецессив-ному типу (РДБЭ). В обоих случаях заболевание развивается вследствие мутации в гене, отвечающем за синтез коллагена VII типа (COL7A1). Буллы при этом образуются между базальной мембраной и дермой [1, 3, 7-9].
Доминантный тип ДБЭ характеризуется наличием сливных буллезных элементов уже при рождении ребенка. Течение в большинстве случаев генерализованное, однако буллы могут локализоваться только на нижних конечностях, локтях или коленях вследствие механической травма-тизации. Рецидивирующее течение приводит к формированию милий, атрофических рубцов, особенно на конечностях, а также ониходистро-фиии и в конечном счете к утрате ногтей. Однако тяжелые вторичные деформации верхних и нижних конечностей, а также внекожные проявления встречаются редко.
Рецидивирующее течение приводит к формированию милий, атрофических рубцов, особенно на конечностях, а также ониходистро-фиии и в конечном счете к утрате ногтей. Однако тяжелые вторичные деформации верхних и нижних конечностей, а также внекожные проявления встречаются редко.
Рецессивный генерализованный тип (ранее Аллопо — Сименса) имеет тяжелое клиническое течение, определяется генерализованное образование пузырей и эрозий с последующим формированием атрофических рубцов, ониходистрофии, с утратой ногтей, а также развитием тяжелых псевдосиндактилий кистей и стоп. Кроме того,
с возрастом развиваются контрактуры локтевых и коленных суставов, кистей и стоп. Вовлечение в процесс слизистой желудочно-кишечного тракта приводит к формированию вторичной микросто-мии, повреждению слизистой пищевода с формированием его рубцовых стенозов и нарушением функции глотания, что в комплексе усугубляет нутритивную недостаточность, вызывает хроническую анемию, задержку роста и остеопороз. У пациентов с данным подтипом крайне высокий риск образования агрессивных плоскоклеточных карцином.
РДБЭ генерализованный, другой (не Аллопо — Сименса): характеризуется локализацией пузырей на руках, ногах, коленях и локтях, иногда на сгибах, на туловище. Заболевание протекает менее тяжело, чем тяжелый генерализованный подтип РДБЭ, заживление происходит без образования рубцов.
Синдром Киндлера является чрезвычайно редким рецессивным генодерматозом, который включает в себя мутации в гене, кодирующем структурный белок КтёНп-1 [7]. Пузыри образуются в любом слое кожи. При синдроме Киндлера имеет место генерализованное повреждение кожных покровов, а также поражение слизистых оболочек желудочно-кишечного тракта, в том числе с образованием анального стеноза. У данных пациентов повышен риск развития плоскоклеточного рака слизистой оболочки полости рта.
Диагностика
Учитывая схожесть клинической картины различных видов БЭ, самой большой ошибкой является попытка в неонатальном периоде определить тип и подтип БЭ без соответствующего тестирования. Даже в пределах одного подтипа, с той же мутацией пациенты могут иметь различную клиническую картину.
В зависимости от периода проведения диагностических мероприятий их можно подразделить на пренатальные и постнатальные.
Биопсия кожи. Исследование биопсии кожи методом трансмиссионной электронной микроскопии и/или иммунофлюоресцентной визуализации антител-антигенов является одним из способов установления диагноза ДБЭ. Следует отметить важность правильного выполнения забора биоптата. Биопсия должна быть взята из переднего края свежего (< 12 часов) или вскрывшегося пузыря, с захватом некоторого количества неизмененной прилегающей кожи, так как претерпевшие изменения волдыри могут давать неясный морфологический диагноз.
Для предварительной диагностики — на каком из уровней кожи произошло повреждение и какие именно белки вовлечены в процесс — в настоящее время используется иммунофлюоресценция. Метод основан на связывании моноклональных антител с белками (антигенами), присутствующими в норме. Если специфические антигены отсутствуют, соответственно, будет отсутствовать окрашивание и выявление недостающих белков, что позволит установить тип, а иногда и подтип БЭ. Например, при ДБЭ окрашивание коллагена VII с использованием антител уменьшается или отсутствует. При легкой форме окрашивание коллагена VII может оказаться нормальным, однако ниже lamina densa и ниже окрашенного коллагена VII можно визуализировать расщепление плоскости структур дермы в виде пузырьков или микропузырьков. В то же время нормальное окрашивание для других антигенов (например, ламинин-332, коллаген XVII, плектин, а6^4-интегрин и кератин-5 и -14) подтверждает диагноз ДБЭ [10, 11].
Тем не менее, особенно при легких формах БЭ, косвенных данных иммунофлюоресцентно-го исследования не достаточно, чтобы поставить диагноз, так как выявляются почти нормальные уровни антигенов и не наблюдается расщепления слоев.
В таких случаях выполняется трансмиссионная электронная микроскопия, которая является «золотым стандартом» для диагностики БЭ. Электронно-микроскопическое исследование биоптата кожи визуализирует структуры зоны базальной мембраны, в частности, может определить количество и морфологию анкерных фибрилл, наличие и морфологию гемидесмосом, кератина промежуточных филаментов. Так, при всех формах ПБЭ расщепление визуализируется на уровне lamina lucida базальной мембраны эпидермиса или чуть выше базальной мембраны, на уровне гемидесмо-сом низкого уровня эпидермиса. При типе Хер-литца определяется значительное уменьшение количества гемидесмосом, их гипоплазия, а также выраженное снижение количества скрепляющих нитей. При типе не-Херлитца гемидесмосомы могут быть гипоплазированы, количество скрепляющих нитей снижено. При некоторых формах ДБЭ в периоде новорожденности коллаген VII типа может задерживаться внутриклеточно в базаль-ном слое эпидермиса вместо того, чтобы перемещаться в зону базальной мембраны.
Генетический анализ. В настоящее время при четырех основных типах БЭ известно до 24 генетических подтипов заболевания. Идентификации типа наследования, анализ структуры ДНК выполняются с целью определения локализации му-
тации, ее типа, что важно для определения дальнейшего прогноза течения заболевания, а также для оценки вероятности рождения ребенка с данным заболеванием у родителей, либо уже имеющих детей с БЭ, либо которые сами имеют данное заболевание. После того, как генетическая мутация идентифицируется в семье, возможно осуществление пренатальной диагностики, начиная с 11 недели беременности путем выполнения амни-оцентеза, биопсии хорионических ворсинок. При выполнении экстракорпорального оплодотворения в семьях с БЭ возможно проведение преимпланта-ционной генетической диагностики [12, 13].
При выполнении экстракорпорального оплодотворения в семьях с БЭ возможно проведение преимпланта-ционной генетической диагностики [12, 13].
Немаловажную роль генетическое картирование играет и в разработке специфичной терапии с использованием препаратов генной инженерии, что в настоящее время является наиболее перспективным методом патогномоничного лечения.
Лечение
Лечение БЭ представляет собой комплекс мероприятий, направленных как на купирование существующих проявлений, так и на предотвращение появления новых элементов.
Свежие везикальные элементы следует вскрывать и дренировать, чтобы предотвратить дальнейшее распространение процесса от давления жидкости [6, 10]. В большинстве случаев повязки в области пузырей состоят из трех слоев. Первый (основной) слой должен свободно прилегать к поверхности кожи, чтобы не производить дополнительную травму и не повреждать верхние слои эпидермиса. Первичные слои могут состоять из повязок, пропитанных смягчающими (сетка на вазелиновой основе) местными антисептическими препаратами (Adaptic®, Xeroform®), обладать антиадгезивными свойствами (например, Telfa® или N-terface®). Также используются силиконовые повязки без липкого слоя (например, Mepitel® или Mepilex®). Для профилактики и лечения инфекции, а также ускорения ранозаживления в основном слое могут использоваться эпителизиру-ющие мази (солкосерил, пантенол), препараты, содержащие оксид цинка и обладающие антисептическими свойствами (антибиотики, серебро). Использование местных глюкокортикоидных препаратов может приводить к ухудшению местного статуса и должно применяться в течение короткого времени в тяжелых случаях БЭ. Второй слой обеспечивает фиксацию основного и составляет многослойность повязки для увеличения активности пациента без механической травматиза-ции. Третий слой обычно обладает эластичными свойствами, что обеспечивает общую целостность
повязки (например, ТиЫ1а81®, СоЬап®). Однако у пациентов с ПБЭ, в отличие от других типов, чрезмерное бинтование может привести к увеличению количества пузырей, по-видимому, в результате повышения местной температуры и потоотделения. Таким пациентам необходимо применять адгезивные повязки с минимальным количеством дополнительных слоев [6].
Однако у пациентов с ПБЭ, в отличие от других типов, чрезмерное бинтование может привести к увеличению количества пузырей, по-видимому, в результате повышения местной температуры и потоотделения. Таким пациентам необходимо применять адгезивные повязки с минимальным количеством дополнительных слоев [6].
Немаловажной проблемой является изготовление ортопедической обуви для пациентов с БЭ, которая должна быть атравматичной и удобной, чтобы предотвратить формирование пузырей. В настоящее время разработана одежда, содержащая серебряные нити, что позволяет уменьшить инфицирование поврежденных поверхностей, в том числе при ходьбе. Все дети с БЭ нуждаются в постоянном реабилитационном лечении, включающем различные виды физиотерапевтических процедур. Адекватное реабилитационное лечение позволяет выполнять эффективную разработку стойких контрактур суставов, что уменьшает количество оперативных вмешательств на верхних и нижних конечностях. Однако закрепление результатов лечения с помощью фиксирующих ортезов представляет значительную проблему у данных детей вследствие повышенного травмирования кожи при жесткой фиксации, этим и обусловлен высокий процент рецидивов деформации [12].
Британская ассоциация дерматологов сообщила о применении инъекций ботулинического токсина в область стоп с целью снижения болевого синдрома у пациентов с обширными буллами подошвенной поверхности. Средняя продолжительность эффекта составляла 3 месяца. Электронно-микроскопическое исследование показало исчезновение внутриэпидермальных расслоений на фоне бутулинотерапии [14]. Однако в настоящее время наиболее широко используется местное применение препаратов алюминия для снижения потоотделения в области стоп.
Хирургическое лечение применяется для коррекции вторичных деформаций (псевдосиндакти-лий), контрактур верхних и нижних конечностей, для закрытия обширных кожных дефектов, в том числе с использованием «гибридных» кожных трансплантатов, содержащих кератиноциты пациента и донорские фибробласты [9, 15]. В настоящее время активно развивается генная терапия БЭ, за которой будущее лечения этой сложной категории больных. Замена дефектного гена на нормально функционирующий — одна из основных целей генной терапии. Таким образом, рецессивный ДБЭ — идеальная модель для генной терапии, так как все его варианты вызваны мута-
В настоящее время активно развивается генная терапия БЭ, за которой будущее лечения этой сложной категории больных. Замена дефектного гена на нормально функционирующий — одна из основных целей генной терапии. Таким образом, рецессивный ДБЭ — идеальная модель для генной терапии, так как все его варианты вызваны мута-
циями в одном гене (COL7A1), кодирующем коллаген VII типа, ключевой компонент крепления фибрилл, соединяющих эпидермис и дерму.
Введение пациентам с РДБЭ мезенхимальных стромальных клеток (МСК) — одно из новых направлений клеточной терапии. Согласно последним исследованиям применение МСК улучшает и ускоряет ранозаживление путем стимуляции производства ангиопротективных факторов, таких как XVII фактор роста эндотелия, а также оказывает опосредованное иммунодепрессивное воздействие путем активации фактора некроза опухоли, который, в свою очередь, уменьшает местные воспалительные реакции. Однако до настоящего момента не изучены механизмы миграции клеток МСК к зоне поражения, а также необходима оценка частоты развития тяжелых побочных эффектов (например, реакции трансплантат против хозяина).
В 3-ю стадию вошло исследование инъекций ‘Т-фибробластов пациентам с дистрофической формой БЭ, проводимое Международной исследовательской ассоциацией помощи детям с ДБЭ (DeBRA). Согласно этим данным подкожное введение фибробластов приводит к появлению новых отложений коллагена VII типа и полной регенерации слоев, ранее пострадавших [15-17].
Несмотря на преимущественно кожные проявления эпидермолиза, коррекция его внекожных проявлений не менее важна. Наличие язвенных элементов в ротовой полости, микростомии, стеноза пищевода, нарушение переваривания и всасывания, с одной стороны, и постоянное увеличение потребности в энергии и питательных веществах — с другой, приводят к замедлению репарации кожных покровов и возникновению воспалительных и инфекционных процессов. Поэтому в комплексном лечении БЭ важную роль играет восполнение белково-энергетической недостаточности, коррекция водно-электролитных нарушений у детей раннего возраста, остеоиндук-тивная терапия (витамин D3, препараты кальция), а также хирургическое лечение вторичных стриктур желудочно-кишечного тракта [18].
Поэтому в комплексном лечении БЭ важную роль играет восполнение белково-энергетической недостаточности, коррекция водно-электролитных нарушений у детей раннего возраста, остеоиндук-тивная терапия (витамин D3, препараты кальция), а также хирургическое лечение вторичных стриктур желудочно-кишечного тракта [18].
Таким образом, не существует единого подхода в диагностике и лечении буллезного эпидермолиза. Сложность дифференциальной диагностики подтипов приводит к запоздалому специфичному лечению и раннему развитию тяжелых осложнений.
Описание клинического случая
Пациент Х. поступил в клинику артрогрипо-за НИДОИ им Г.И. Турнера в возрасте 5 месяцев из детского дома с направляющим диагнозом:
«Рецессивный дистрофический буллезный эпидер-молиз. Артрогрипоз с поражением верхних и нижних конечностей». Из анамнеза известно, что ребенок от IV беременности (1-3 девочки здоровы) близкородственного брака, роды в срок в головном предлежании. При рождении у ребенка отмечались сгибательные контрактуры правого локтевого и обоих коленных суставов до 90°, эквино-вальгусная деформация обеих стоп, на основании чего ребенку поставлен диагноз: «Артрогрипоз с поражением верхних и нижних конечностей». При этом в области ладонных поверхностей определялись обширные мацерации по типу «перчатки». На 9-е сутки жизни ребенок переведен для дальнейшего лечения в ДГБ № 1. В динамике в области стоп и кистей образовались эрозии с признаками инфицирования, в связи с чем ребенок получал местное консервативное лечение. Мальчик консультирован дерматологами СПбГПМА — поставлен диагноз: «Рецессивный дистрофический буллезный эпидермолиз». При выписке из стационара нижние конечности были фиксированы пластиковыми ортезами для фиксации стоп в максимально возможном правильном положении, однако выраженная травматизация кожных покровов не позволила ребенку носить их длительное время. В отделение артрогрипоза ребенок поступил для консервативного лечения деформаций нижних конечностей. При клиническом осмотре в области кистей визуализировались множественные буллезные элементы неправильной формы: свежие в области правой кисти и вскрывшиеся в области левой, без выраженных воспалительных проявлений (рис. 2). Также определялась сгибательная контрактура правого локтевого сустава до 130°.
В отделение артрогрипоза ребенок поступил для консервативного лечения деформаций нижних конечностей. При клиническом осмотре в области кистей визуализировались множественные буллезные элементы неправильной формы: свежие в области правой кисти и вскрывшиеся в области левой, без выраженных воспалительных проявлений (рис. 2). Также определялась сгибательная контрактура правого локтевого сустава до 130°.
В области нижних конечностей: выявлены сгибательно-приводящие контрактуры тазобедренных суставов, сгибательные контрактуры коленных суставов до 140° (рис. 3).
Стопы имели стойкое эквино-вальгусное положение с максимальной тыльной флексией до 20°. Первые пальцы обеих стоп также имели сгибатель-но-приводящие контрактуры (рис. 4).
По данным рентгенографии выявлялось отведение переднего отдела стопы и пронация заднего (рис. 5).
В области пальцев стоп определялись вскрывшиеся буллезные элементы до 1,5 х 0,5 см с умеренной гиперемией. На подошвенной поверхности в области пятки левой стопы визуализировался обширный поверхностный везикальный элемент в стадии эпителизаии.
Учитывая наличие открытых раневых поверхностей в области стоп, лечение было решено на-
Рис. 2. Внешний вид кистей при поступлении
ча
Рис. 3. Сгибательная контрактура коленных суставов. Внешний вид при поступлении
111
Рис. 4. Внешний вид стоп при поступлении
А
л.1 . Г А
Рис.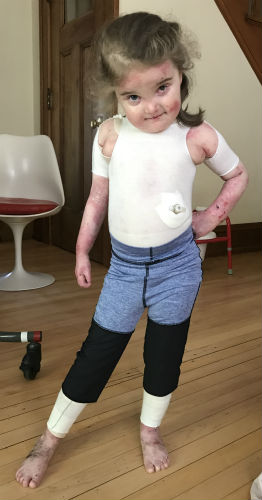 5. Рентгенография стоп при поступлении (боковая и прямая проекции)
5. Рентгенография стоп при поступлении (боковая и прямая проекции)
чать с наложения многослойных повязок с рано-заживляющими мазями, в том числе с препаратами цинка. Назначены курсы физиотерапевтического лечения в виде поляризованного света на участки деэпителизированных тканей. После стабилизации кожных проявлений проводилось этапное гипсование эквино-вальгусной деформации стоп с ахиллотомией с двух сторон. Проводилась ну-тритивная поддержка белковыми препаратами, а также интенсивная терапия микроэлементами, содержащими оксид цинка, ионы кальция, фосфора и марганца. В комплексную терапию входил витамин Б3 в возрастных дозировках.
Наличие сопутствующего БЭ обусловило необходимость более частой смены гипсовых повязок, на первых этапах она производилась каждые четыре дня, так как при увеличении длительности иммобилизации резко ухудшалось состояние кожных покровов: увеличивалось количество свежих
буллезных элементов и формировались участки обширных мацераций. Таким образом, для полной коррекции положения стоп понадобилось 8 гипсовых повязок (рис. 6).
Сгибательные контрактуры правого локтевого и коленных суставов тоже требуют этапного гипсования. Однако, по-нашему мнению, его целесообразно проводить в более позднем возрасте, так как выраженная белково-нутритивная недостаточность сама по себе способствует образованию новых буллезных элементов даже без механического воздействия. В то же время необходимость длительного ношения гипсовых повязок будет увеличивать риск формирования обширных эпидер-мальных дефектов и, как следствие, развития генерализованного инфицирования кожных покровов. Следует отметить, что лечение сгибательных контрактур коленных суставов возможно только после стойкой коррекции деформации стоп.
Рис. 6. Результат этапного лечения вальгусного положения стоп
6. Результат этапного лечения вальгусного положения стоп
Обсуждение
Как уже отмечалось, серьезной проблемой является дальнейшее ортезирование пациента для предотвращения рецидива деформации. Данная ситуация усугубляется наличием сопутствующего артрогрипоза, при котором ношение жестких ор-тезов не исключает возвращение стойкой деформации. Даже самые современные фиксирующие средства, к сожалению, не рассчитаны на сочетание столь сложных заболеваний.
Заключение
Таким образом, все дети с подозрением на бул-лезный эпидермолиз должны проходить точную верификацию диагноза с помощью генетического картирования для определения оптимальной тактики дальнейшего лечения и общего прогноза. Сочетание тяжелой формы буллезного эпидер-молиза и артрогрипоза требует особого подхода к консервативному и оперативному лечению данной группы пациентов, делает необходимой тщательную подготовку к выполнению этапных гипсований и других консервативных мероприятий. Создание нового поколения фиксирующих орте-зов и разработка плана индивидуальных реабилитационных программ должны улучшить качество жизни пациентов и их социальную адаптацию.
Список литературы
1. Intong LR, Murrell DF. Inherited epidermolysis bullosa: new diagnostic criteria and classification. Clin Dermatol. 2012;30:70-7. doi: 10.1016/j.clindermatol.2011.03.012.
2. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa. J Am Acad Dermatol. 2009;61:387-402.
doi: 10.1016/j.jaad.2009.03.053.
3. Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-950. doi: 10.1016/j.jaad.2008.02.004.
The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-950. doi: 10.1016/j.jaad.2008.02.004.
4. Bolling MC, Lemmink HH, Jansen GH, Jonkman MF. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75 % of the patients. Br J Dermatol. 2011;164:637-44.
doi: 10.1111/j.1365-2133.2010.10146.x.
5. Pfendner EG, Bruckner AL. Epidermolysis Bullosa Simplex. Initial Posting: October 7, 1998; Last Update: September 1, 2011. doi: 10.1007/ springerreference_35076.
6. Pope E, Lara-Corrales I, Mellerio J, et al. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol. 2012;67:904-17.
doi: 10.1016/j.jaad.2012.01.016.
7. Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70:1103-26. doi: 10.1016/j.jaad.2014.01.903.
8. Murrell D. Epidermolysis Bullosa: Part I — Pathogenesis and Clinical Features. 1 ed. Vol. 28-1. Dermatologic Clinics. Elsevier, 2010. doi: 10.1016/j.det.2009.10.020.
9. Woodley DT, Chen M. Recessive Dystrophic Epidermolysis Bullosa: Advances in the laboratory leading to new therapies. J of Investigative Dermatology. 2015;135:1705-1707. doi: 10.1038/jid.2015.149.
10. Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28:107-114. doi: 10.1016/j.det.2009.10.020.
11. Soro L, Bartus C, Purcell S. Recessive Dystrophic Epidermolysis Bullosa: A eview of disease: Pathogenesis and update on future therapies. J Clin Aesthet Dermatol. 2015;8(5):41-46. Available from: http://www.ncbi.nlm. nih.gov/pmc/articles/PMC4445895/
Soro L, Bartus C, Purcell S. Recessive Dystrophic Epidermolysis Bullosa: A eview of disease: Pathogenesis and update on future therapies. J Clin Aesthet Dermatol. 2015;8(5):41-46. Available from: http://www.ncbi.nlm. nih.gov/pmc/articles/PMC4445895/
12. DEBRA International. Available from: http://www. debra-international.org/debra.html
13. Liu N, Guo H, Kong X, Shi H, et al. COL7A1 gene mutation analysis of dystrophic epidermolysis bullosa and prenatal diagnosis. Exp Dermatol. 2015;95(4):277-82. PMID: 25877244.
14. Swartling C, Karlqvist M, Hymnelius K, et al. Botulinum toxin in the treatment of sweat-worsened foot problems in patients with epidermolysis bullosa simplex and pachyonychia congenita. Br J Dermatol. 2010;163(5):1072-6.
doi: 10.1111/j.1365-2133.2010.09927.x.
15. Wong T, Gammon L, Liu L, et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2008;128:2179-89. doi: 10.1038/jid.2008.78.
16. Woodley DT, Wang X, Amir M, et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133:1910-3. doi: 10.1038/jid.2013.10.
17. Hovnanian A. Systemic protein therapy for recessive dystrophic epidermolysis bullosa: how far are we from clinical translation? J Invest Dermatol. 2013;133(7):1719-21. doi: 10.1038/jid.2013.137.
18. Zidorio APC, Dutra ES, Leäo DOD, Costa IMC. Nutritional aspects of children and adolescents with epider-molysis bullosa: literature review. An Bras Dermatol. 2015;90(2):217-23. doi: 10.1038/jid.2013.137.
2015;90(2):217-23. doi: 10.1038/jid.2013.137.
DYSTROPHIC EPIDERMOLYSIS BULLOSA ASSOCIATED WITH CONGENITAL CONTRACTURES OF THE UPPER AND LOWER LIMBS: LITERATURE REVIEW
Agranovich O.E., Buklaev D.S., Tikhonenko T.I.
The Turner Scientific and Research Institute for Children’s Orthopedics, Saint-Petersburg, Russian Federation
Epidermolysis bullosa (EB) is a rare hereditary disease. Its main feature is vesication and weeping sores (erosions) of the skin and mucous membranes, resulting from a minor injury. Clinical manifestations of the disease may vary from localized vesicles on the hands and feet to a generalized rash of the skin as well as lesions of the mucosa of the inner organs. At present, there are four main groups of EB: simple, intermediate, dystrophic, and Kindler syndrome. Mutations cause changes in the structure of the proteins responsible for the adhesion between layers of the dermis, leading to vesication. Treatment of EB is a challenge because
of the lack of opportunities for the direct influence on the disease process, and its main purpose is to correct the existing cutaneous manifestations and prevent the occurrence of new elements. This article describes the main types of EB, methods of current diagnosis, and treatment of the disease as well as a clinical case of a rare combination of two severe disorders: 1) dystrophic EB and 2) arthrogryposis with upper and lower limb involvement.
Keywords: Epidermolysis bullosa, arthrogryposis, flexion contractures of extremities.
Сведения об авторах
Агранович Ольга Евгеньевна — д. м. н., руководитель отделения артрогрипоза ФГБУ «НИДОИ им. Г.И. Турнера» Минздрава России. E-mail: [email protected].
Г.И. Турнера» Минздрава России. E-mail: [email protected].
Буклаев Дмитрий Степанович — к. м. н., заведующий отделением артрогрипоза ФГБУ «НИДОИ им. Г.И. Тур-нера» Минздрава России. E-mail: [email protected].
Тихоненко Татьяна Ивановна — к. м. н., ведущий научный сотрудник отделения артрогрипоза ФГБУ «НИДОИ им. Г.И. Турнера» Минздрава России. E-mail: [email protected].
Agranovich Olga Evgenievna — MD, PhD, professor, head of the department of arthrogryposis. The Turner Scientific and Research Institute for Children’s Orthopedics. E-mail: [email protected].
Buklaev Dmitry Stepanovich — MD, PhD, chief of the department of arthrogryposis. The Turner Scientific and Research Institute for Children’s Orthopedics. E-mail: [email protected].
Tikhonenko Tatiana Ivanovna — MD, PhD, leading research associate of the department of arthrogryposis. The Turner Scientific and Research Institute for Children’s Orthopedics. E-mail: [email protected].
как продвинулось лечение буллёзного эпидермолиза в России
Во многих медучреждениях России принято считать, что от кожных болезней никто не умирает, поэтому оперативной помощи пациентам с буллёзным эпидермолизом не оказывается, утверждает Николай Мурашкин, заведующий отделением дерматологии Национального медицинского исследовательского центра (НМИЦ) здоровья детей Минздрава России.
В научном центре здоровья детей работает единственное отделение для «детей-бабочек» в России. Здесь оказывают квалифицированную помощь детям с буллёзным эпидермолизом. Однако, несмотря на редкость патологии, в нашей стране крайне не хватает специалистов на местах, которые бы смогли быстро поставить диагноз и назначить лечение.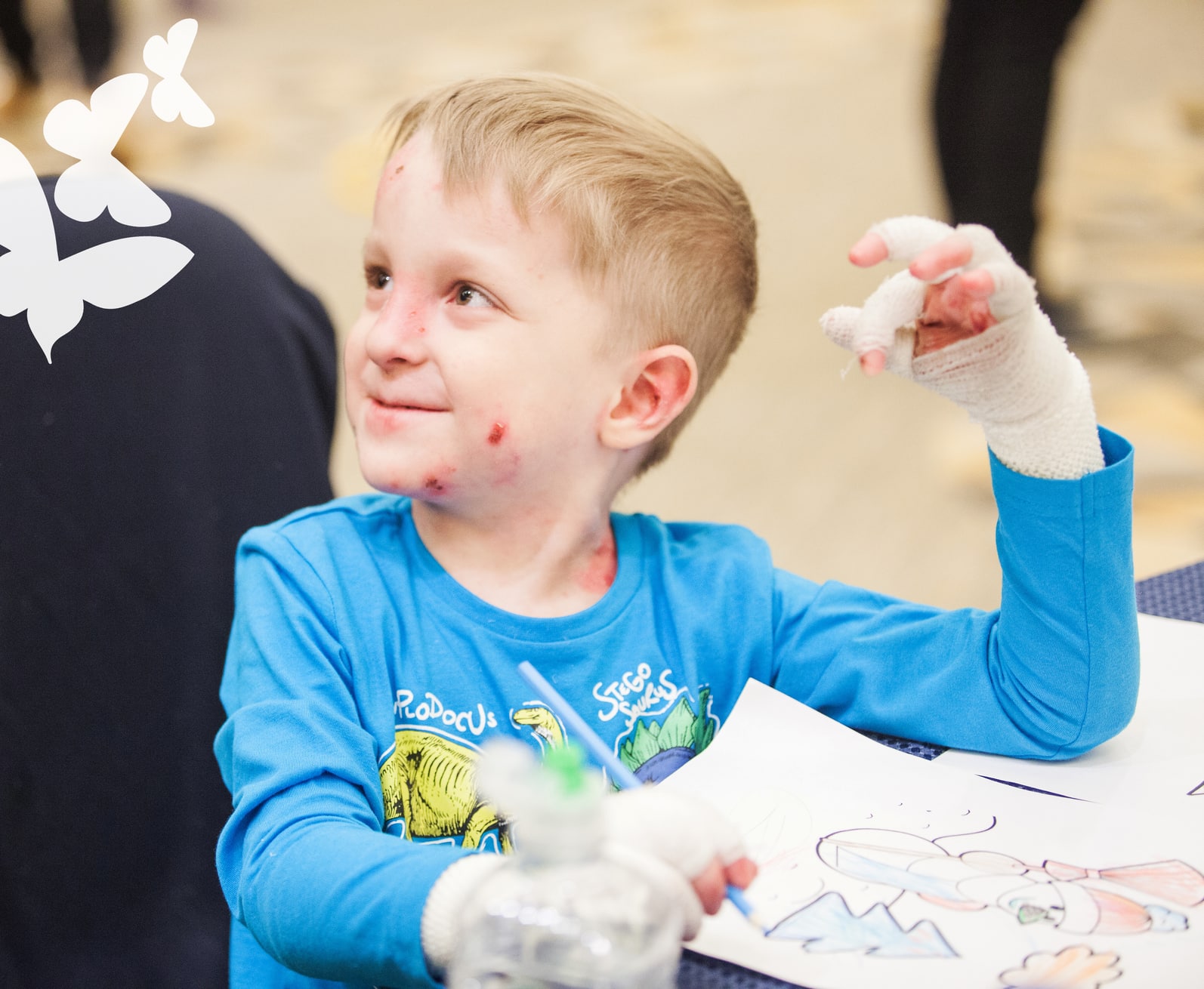
«К большому сожалению, кадровый голод испытывается на многих территориях России. Поскольку заболевание очень редкое и мало изучено специалистами, отсутствует слаженная система организации помощи таким детям, — рассказал Николай Мурашкин, выступая на паблик-токе фонда «Дети-бабочки» в центре «Благосфера». — Это заболевание затрагивает основу соединительной ткани, на которой крепится вся кожа. Для помощи таким детям не хватает одного педиатра и дерматолога, необходима большая команда специалистов. Неправильно, что сегодня в России проблема помощи детям отдана кожно-венерологическим диспансерам. В диспансерах можно провести далеко не все исследования пациента. У нас в 2/3 регионов помощь таким детям находится в неудовлетворительном состоянии».
При этом, по словам Мурашкина, российские медики во время изучения данного заболевания смогли сделать уникальные наблюдения, которых нет в мировой практике. В России выявлены новые формы буллёзного эпидермолиза, которые имеют мутации и отличаются от европейских.
«Сейчас генная инженерия далеко продвинулась в исследованиях. И препараты генной инженерии для многих редких заболеваний стоят на второй-третьей стадиях клинических испытаний. Они вот-вот войдут в клиническую практику, — заявила Юлия Коталевская, заведующая консультативным отделением медико-генетического центра МОНИКИ, врач-генетик. — Научные группы открывают все новые препараты. Конечно, через два-три года это не будет массовой панацеей, но итог будет все равно положительный. В России тоже есть возможности заниматься подобного рода исследованиями. Но, когда обмениваемся данными, получается, что Россия для европейцев остается загадкой с точки зрения этой болезни. У нас множество национальностей и поэтому возникают различные специфики генетических заболеваний».
Кроме того, сегодня встает вопрос о помощи не только детям, но и взрослым с буллезным эпидермолизом. «Сейчас благодаря нашей работе, благодаря тому, что врачи больше знают о заболевании, мы быстрее реагируем, к нам обращаются врачи и мамы, качество и длительность жизни «бабочек» имеет тенденцию к улучшению и увеличению. Те, кто не доживал до подросткового возраста, теперь перешагивает рубеж совершеннолетия и живет дальше, – рассказала актриса и попечитель фонда Ксения Раппопорт. – Но эта невероятная радость одновременно тяжелое, сложное и на сегодняшний день совершенно неподъемное для нас бремя. У нас ушли годы на то, чтобы внедриться и понять, как организовать помощь в детской медицине. Но мы столкнулись с проблемой. Ребёнок, например, в фонде с 10 лет, он вырос, ему исполнилось 18, и что? У него прошел буллёзный эпидермолиз? Нет. Ему стало легче? Нет. И мы ему говорим: живи как можешь? Это невозможно. А с другой стороны – невозможно. Потому что у нас нет ресурсов. И помогать выборочно мы не можем. Только в Дагестане, например, взрослых больных 110 человек».
Ксения Раппопорт. Фото предоставлено фондом «Дети-бабочки»
Ранее сообщалось, что пациенты с редкими заболеваниями из программы «Семь нозологий» будут обеспечены лекарственными препаратами российского производства.
«В этом году заканчиваются клинические исследования препаратов для лечения муковисцидоза и болезни Гоше. Таким образом, пациентов с орфанными заболеваниями из программы «Семь нозологий» получится обеспечить российскими препаратами в рамках тех инициатив, которые звучали в госпрограмме «Фарма 2020», — сообщил Дмитрий Кудлай, представитель Ассоциации фармпроизводителей ЕАЭС. По его словам, из этих 24 орфанных заболеваний для 18 лекарственные препараты уже в разработке, из них 12 — на той или иной стадии клинических исследований.
Как сообщает РИА Новости, в настоящее время в России больные редкими заболеваниями обеспечиваются лекарственными препаратами из двух источников: федерального (программа «7 высокозатратных нозологий» — гипофизарный нанизм, гемофилия, муковисцидоз, болезнь Гоше) и регионального (24 нозологии, финансируется из бюджетов регионов).
По данным Минздрава РФ, в федеральном регистре лиц с редкими заболеваниями, в настоящее время находятся 17 226 человек, в том числе 8 762 ребенка.
Буллёзный эпидермолиз — группа генетически и клинически гетерогенных заболеваний, характеризующаяся образованием пузырей и эрозий на коже и слизистых оболочках, ранимостью кожи и ее чувствительностью к незначительной механической травме.
Больше новостей некоммерческого сектора в телеграм-канале АСИ. Подписывайтесь.
Современные особенности клиники, диагностики и терапии больных буллезным эпидермолизом | #01/18
Буллезный эпидермолиз (БЭ) — это группа редких наследственных генетических заболеваний кожи, обусловленных мутациями ряда генов, ответственных за синтез структурных белков кожи. Для заболевания характерна склонность кожи и слизистых оболочек к образованию пузырей, преимущественно на местах незначительного механического воздействия, вследствие нарушения межклеточных связей в эпидермисе или дермоэпидермальном соединении [1, 2]. Наиболее часто пациенты с буллезным эпидермолизом регистрируются в возрасте от 1 до 5 лет. По данным Ю. Ю. Коталевской с соавт., на долю дистрофического варианта приходится больше половины пациентов от общего количества всех форм буллезного эпидермолиза [3]. На сегодняшний день своевременная диагностика и терапия буллезного эпидермолиза остаются одной из главных проблем в медицине. При любой форме буллезный эпидермолиз протекает тяжело, инвалидизация наступает в первые годы течения болезни.
Развитие буллезного эпидермолиза обусловлено мутациями генов, кодирующих структурные белки кожи, которые обеспечивают связь между эпидермисом и дермой. К настоящему времени в 15 генах структурных белков кожи выявлено более 1000 мутаций, способных приводить к развитию различных клинических типов врожденного буллезного эпидермолиза [4–6]. С мутациями связаны нарушения синтеза белков: отсутствие белка, синтез функционально неполноценного белка, синтез белка с нарушениями структуры, облегчающими доступ к белку протеаз, что приводит к его быстрому разрушению. Белками, с которыми связано развитие заболевания, являются кератины 5 и 14, десмоплакин, плакофилин-1, плектин, интегрин α6β4, ламинин 332, коллагены VII и XVII типов, киндлин. Эти белки имеют различную локализацию в коже: в кератиноцитах локализуются кератины 5 и 14, внутри светлой пластинки (lamina lucida) базальной мембраны — интегрин α6β4, ламинин 332, коллаген XVII типа, под темной пластинкой (lamina densa) базальной мембраны — коллаген VII типа, на разных уровнях эпидермиса — киндлин [4, 5].
Выделяют несколько основных типов буллезного эпидермолиза на основе особенностей механизма образования пузыря и клинической картины: простой буллезный эпидермолиз, пограничный буллезный эпидермолиз, дистрофический буллезный эпидермолиз, синдром Киндлера (разный уровень образования пузырей) [7].
Эпидермолиз буллезный простой
Эпидермолиз буллезный простой характеризуется образованием внутриэпидермальных пузырей в результате дезинтеграции и цитолиза кератиноцитов без признаков рубцевания, атрофии и образования милиумов. Тип наследования аутосомно-доминантный. Первые признаки заболевания обычно проявляются на первом году жизни, иногда могут быть уже при рождении ребенка. На месте легкой травматизации, чаще в области кистей, стоп, спины, локтевых и коленных суставов, затылочной области, на неизмененной коже появляются пузыри различных размеров (от 0,5 до 7 см и более) с плотной покрышкой и прозрачным содержимым. Симптом Никольского отрицательный, акантолитические клетки в содержимом пузыря отсутствуют. Через несколько днем пузыри вскрываются, образуя эрозии, покрывающиеся корками и быстро эпителизирующиеся, не оставляя рубцовых изменении кожи или атрофии. Пузырей обычно больше в теплый период года при выраженном гипергидрозе. С возрастом поражения локализуются в основном на конечностях, особенно на стопах и кистях, чему способствует большая травматизация этих участков кожи, тесная, плохо подобранная обувь, а также на участках тесного прилегания одежды. Пузыри появляются на протяжении всей жизни, но в постпубертатный период их количество уменьшается. Слизистые оболочки, ногти не поражаются или их изменения минимальны. Общее состояние больного не изменяется. Возможна пренатальная диагностика этой формы заболевания по высокому содержанию в сыворотке крови беременной α-фетопротеина во II триместре. Эпидермолиз буллезный простой летний Вебера–Коккейна — абортивная локализованная форма эпидермолиза буллезного простого. Характеризуется образованием пузырей на коже кистей и стоп лишь в летнее время года при выраженном ладонно-подошвенном гипергидрозе [8, 12].
Эпидермолиз буллезный соединительный
Эпидермолиз буллезный соединительный характеризуется образованием подэпидермальных пузырей за счет поражения lamina lucida эпидермодермального соединения, расположенной между плазматической мембраной базальных кератиноцитов и базальной мембраной кожи, и развитием атрофических изменений кожи в очагах поражения. Возможна пренатальная диагностика с помощью биопсии кожи 18-недельного плода на основании выявления указанных изменений. Тип наследования аутосомно-рецессивный.
Процесс характеризуется появлением пузырей и эрозий уже при рождении ребенка или вскоре после него. В течение нескольких дней процесс генерализуется. Основная локализация высыпаний — кожа груди, головы, слизистые оболочки рта, гортани, трахеи. Хотя кожа кистей и стоп не изменена, ногтевые пластинки дистрофичны, развиваются анонихия, акроостеолиз. Образующиеся на месте пузырей эрозивные поверхности заживают медленно, оставляя участки атрофии кожи. Рубцов и милиумов нет. Многие дети умирают в первые месяцы жизни от сепсиса, анемии [8].
Эпидермолиз буллезный дистрофический
Эпидермолиз буллезный дистрофический характеризуется образованием пузырей вследствие дерматолиза — гибели коллагеновых фибрилл в сосочковом слое дермы ниже lamina densa. Формируются эрозивно-язвенные поверхности, заживающие рубцами, характерны также образование милиумов, изменение ногтей, волос, зубов и другие аномалии.
Эпидермолиз буллезный дистрофический рецессивный генерализованный
Эпидермолиз буллезный дистрофический рецессивный генерализованный (эпидермолиз буллезный дистрофический полидиспластический) отличается образованием пузырей в сосочковом слое дермы и в результате дерматолиза — лизиса коллагеновых фибрилл с фагоцитозом их макрофагами и разрушениями ниже lamina densa. Патологический процесс связывают с увеличением уровня и активности фермента коллагеназы, разрушающей основной компонент опорных коллагеновых фибрилл — коллаген VII (коллагенолиз). Возможна пренатальная диагностика болезни по результатам биопсии кожи плода на 21-й неделе развития и выявления описанных ранее изменений. Первые признаки заболевания появляются уже при рождении (60% больных) или в первые недели жизни. Крупные пузыри, нередко с гемморрагическим содержимым, возникают спонтанно на любом участке кожного покрова и слизистых оболочек. Обширные длительно не заживающие эрозивно-язвенные поверхности, образующиеся при их вскрытии, затрудняют уход и вскармливание новорожденных. Симптом эпидермальной отслойки положительный. На эрозивно-язвенных, нередко кровоточащих, болезненных участках развиваются вегетации. Заживление их происходит медленно, с формированием уродствующих атрофических рубцов. Рубцовые изменения пищевода, глотки, слизистой оболочки рта могут затруднять прием пищи, облитерировать выводные протоки слюнных желез, ограничивать подвижность языка и привести к развитию лейкоплакии. Поражения глаз в виде эрозивно-язвенного кератита с последующим рубцеванием приводят к потере зрения, рубцовому эктропиону, облитерации протоков слезных желез. Наблюдаются также акроцианоз, склеродермоподобные изменения кожи кистей, стоп с формированием сгибательных контрактур суставов, акроостеолизом и характерной деформацией кистей по типу «варежки» в результате срастания и деформации пальцев. Характерна также дистрофия ногтей, волос, зубов. Возможны нарушения эндокринной (гипофункция щитовидной железы, гипофиза), нервной (эпилепсия, отставание умственного развития) систем. Отмечается высокая летальность в раннем детском возрасте от сепсиса, анемии, нарушения питания, в более старшем возрасте — от злокачественных новообразований кожи, пищевода, органов полости рта [8].
Эпидермолиз буллезный дистрофический доминантный
Эпидермолиз буллезный дистрофический доминантный (эпидермолиз буллезный дистрофический гиперпластический) характеризуется образованием пузырей в дерме (дерматолиз) ниже lamina densa за счет гибели опорных коллагеновых фибрилл; возможна пренатальная диагностика (по аналогии с дистрофическим полидиспластическим буллезным эпидермолизом). Тип наследования аутосомно-доминантный. Первые проявления болезни появляются в раннем детском возрасте или несколько позднее (4–10 лет). Пузыри возникают после незначительной травмы, чаще в области конечностей. Они напряженные, плотные, с серозным или геморрагическим содержимым: вскрываясь, образуют эрозивно-язвенные поверхности, заживающие медленно с образованием мягкой или келоидоподобной рубцовой атрофии вначале розового, затем белого цвета. В области суставов на месте пузырей формируются обширные поля поражения в виде рубцовой ткани с множеством эпидермальных кист (милиумы). Симптом эпидермальной отслойки положительный. Ногти, вовлеченные в процесс, утолщены, дистрофичны. Слизистые оболочки поражаются редко. Волосы, зубы и общее развитие обычно не изменяются, однако часто отмечается ассоциация с ихтиозом, фолликулярным кератозом, гипертрихозом. Диагноз буллезного эпидермолиза основывается на клинических и гистологических данных. Возможна пренатальная диагностика заболевания. Дифференциальный диагноз в раннем детском возрасте проводят с эпидермолитическим ихтиозом, при котором доминирует кератоз; эпидемической пузырчаткой новорожденных, для которой характерно острое начало с лихорадкой, интоксикацией и воспалительными пузырями в результате некротических процессов в эпидермисе, вызванных стафилококком. У детей более старшего возраста некоторые формы БЭ дифференцируют от доброкачественного буллезного пемфигоида, который отличается линеарным отложением IgА вдоль назальной мембраны. Антитела к lamina densa, VII типу коллагена, пемфигоидные и др. помогают установить характер дефекта и уточнить диагностику [8, 9].
На основании только анамнестических и клинических данных не всегда представляется возможным установить диагноз БЭ. У детей старшего возраста в диагностике помогает сбор анамнеза (начало заболевания, значение механического фактора в развитии пузырей и эрозий, постоянно прогрессирующее течение, последовательность развития симптомов, наследственная отягощенность). Обнаружение определенных специфических клинических признаков некоторых форм БЭ может указать на данную патологию, но в большинстве случаев для уточнения диагноза требуется проведение лабораторной диагностики. Традиционное патоморфологическое исследование биоптата кожи не позволяет различить клинические формы БЭ между собой, но при определенном опыте можно отличить по уровню образования пузыря от истинной акантолитической пузырчатки. После исключения всех других возможных заболеваний для подтверждения диагноза заподозренного БЭ и установления его группы могут быть использованы дополнительные методы диагностики: иммунофлюоресцентное антигенное картирование (ИАК), трансмиссионная электронная микроскопия (ТЭМ) и генетический анализ (молекулярная или ДНК-диагностика) [9].
Трансмиссионная электронная микроскопия
До начала использования в практике иммунофлюоресцентного анализа ТЭМ являлась «золотым» стандартом в диагностике БЭ, так как позволяла определить уровень образования пузыря и обнаружить ультраструктурные изменения в коже и непосредственно в поврежденных структурах, таких как кератиновые филаменты, десмосомы, полудесмосомы, крепящие (якорные) филаменты, крепящие (якорные) фибриллы, которые при определенных формах БЭ могут отсутствовать, либо содержаться в коже в недостаточном количестве, либо иметь дефектную структуру [9].
Иммунофлюоресцентное антигенное картирование
Иммунофлюоресцентное антигенное картирование было впервые описано в 1981 г. В основе метода лежит последовательная обработка препарата кожи пациента первичными антителами к структурным белкам кожи и вторичными антителами, мечеными флюоресцирующей меткой, которые связываются с первичными антителами. В связи с тем, что известно большое количество белков, дефекты которых приводят к развитию БЭ (кератины 5 и 14, плектин, плакофилин-1, десмоплакин, ламинин 332, интегрин α6β4, коллагены VII и XVII типов, киндлин-1), необходимо определять экспрессию каждого из них. Поэтому для проведения исследования методом ИАК из полученного биоптата кожи изготавливают несколько гистологических препаратов [9–11].
Метод используется для определения уровня образования пузыря (внутриэпидермально, внутри светлой пластинки базальной мембраны, под плотной пластинкой базальной мембраны). ИАК позволяет также определить, дефицит какого из связывающих структуры дермы и эпидермиса белков наблюдается. ИАК определяет экспрессию структурных белков в зоне дермоэпидермального соединения, то есть их присутствие, снижение или отсутствие их экспрессии. Для исследований методом ИАК используют первичные и вторичные моноклональные и поликлональные антитела. Этот метод диагностики более распространен, легче выполним и дешевле по сравнению с ТЭМ. Для правильной постановки диагноза методом ИАК нужно иметь достаточный опыт получения биообразцов кожи, причем одним из требований к получению биообразцов является наличие свежего (не более 24 часов) пузыря на коже. Пузырь может быть как появившимся спонтанно, так и индуцированным намеренно, например, с помощью трения кожи. Биопсию проводят на границе видимо здоровой кожи и свежего пузыря или в зоне трения (через 30 мин после его окончания). Полученный биоптат сразу же подвергается заморозке жидким азотом или помещается в физиологический раствор (0,9% NaCl) на срок не более 24 часов. Для более продолжительного хранения используют специальную транспортную среду Michel. В таком состоянии биообразцы кожи могут сохраняться в течение нескольких недель [12].
Генетический анализ (молекулярная или ДНК-диагностика)
Генетический анализ (молекулярная или ДНК-диагностика) является оптимальным методом для определения типа наследования и специфических мутаций, имеющихся у больных БЭ, а также наиболее точным методом для верификации различных клинических форм простого, пограничного и дистрофического БЭ. Поиск известных мутаций является первичным диагностическим подходом, особенно в семьях больных БЭ с аутосомно-доминантным типом наследования, в силу того, что данные мутации, как правило, идентичны в семье, страдающей данным заболеванием. Также имеются данные, что поиск неизвестных мутаций может быть осуществлен с использованием ДНК-амплификационного метода и секвенированием РНК дефектных генов [9].
Пренатальная диагностика
Как известно, до настоящего времени не разработано эффективных методов терапии БЭ, поэтому особенно важное значение на стадии планирования семьи, находящейся в группе риска наследования данной патологии, приобретает пренатальная диагностика и генетическое консультирование. Первое сообщение о пренатальной диагностике одной из наиболее тяжелых форм БЭ (летального генерализованного БЭ Герлитца) было опубликовано еще в 1980 г. [13].
Усовершенствование молекулярных методов диагностики позволило проводить пренатальную диагностику на основании анализа фетальной ДНК, которую можно получить из амниотической жидкости и/или ворсинок хориона. Биопсия ворсин хориона и амниоцентез позволяют существенно снизить риск преждевременного прерывания беременности по сравнению с фетоскопией, а также провести данный вид диагностики в I триместре (до 11 недель беременности). Дородовая диагностика возможна только в том случае, когда ее планируют заранее. До наступления беременности нужно установить точно генетический дефект у болеющего члена семьи. Если дефектный ген уже известен, то поиск такой же поломки у плода занимает всего несколько дней, что создает возможность проведения аборта в безопасные для беременной женщины сроки. Трудностью в диагностике может являться мозаицизм, при котором у ребенка могут присутствовать две и более генетически различные популяции клеток [14, 15].
Преимплантационная генетическая диагностика
Преимплантационная генетическая диагностика является перспективным альтернативным методом наряду с вышеперечисленными, кроме того, метод может быть частью экстракорпорального оплодотворения, выявляя тем самым различные отклонения развития эмбриона еще на стадии бластоцисты. Суть данного метода заключается в оплодотворении овоцита (незрелой яйцеклетки) in vitro, что ведет к образованию эмбриона. Когда эмбрион достигает стадии 8- или 12-клеточного субъекта, производится отщепление одной клетки для дальнейшего генетического анализа. Если генетический анализ указывает на нормальный генотип — эмбрион подсаживают в матку, иначе его утилизируют [16].
Неинвазивные методы пренатальной диагностики
Все вышеперечисленные методы пренатальной диагностики являются инвазивными и в той или иной степени могут повлиять на развитие плода и на течение беременности. В связи с этим идет постоянный поиск новых методов, снижающих риск инвазивного воздействия [9].
Среди таких методов можно выделить метод ультразвуковой диагностики, который позволяет визуализировать некоторые особенности развития плода при ограниченном количестве тяжелых патологий, а также другие структуры содержимого матки, которые могут быть идентификаторами некоторых врожденных заболеваний. К таким показателям можно отнести особенности фенотипа плодного яйца: размеры носовой кости, воротниковой области, желточного мешка [9].
Генетическое консультирование лучше проводить дерматологом или генетиком, специализирующимся на БЭ. В конечном итоге диагноз ставится на основании клинического фенотипа, способа наследования, а также, если возможно, провести мутационный анализ пробанда.
В периоде новорожденности наблюдение и симптоматическая терапия за больными врожденным буллезным эпидермолизом (ВБЭ) проводится в условиях отделения интенсивной терапии педиатрического стационара. Основные принципы терапии не отличаются от таковых у взрослых лиц. Лекарственная терапия проводится с учетом возрастных ограничений к назначению лекарственных препаратов. Профилактические прививки противопоказаны только в период нарушения общего состояния ребенка. Беременность у больных ВБЭ обычно протекает без осложнений. При планировании беременности необходимо провести лечение сопутствующих заболеваний, в том числе очагов хронической инфекции, хирургическое устранение контрактур и псевдосиндактилии. Во избежание травмирования принимают меры предосторожности при взятии крови, вагинальном исследовании, пальпации, УЗИ. Основным в период беременности является наружное лечение [7].
Больным буллезным эпидермолизом необходимо избегать физических нагрузок, связанных с повышением потоотделения, травмоопасных ситуаций, резких движений. Перевязочные материалы, одежда, закрытая обувь позволяют свести к минимуму травмирование кожи. При хорошем самочувствии и отсутствии высыпаний на коже допустимо плавание.
Тяжелые подтипы БЭ, а также нетяжелые, но протекающие с поражением полости рта, требуют особого внимания к питанию. Диета должна быть механически, термически и химически щадящей (протертой и полужидкой, не горячей). Питание особенно важно для больных с большой площадью поражения кожи, так как они теряют питательные вещества и влагу с тканевой жидкостью, необходимой для заживления ран и борьбы с инфицированием. Питание больных должно быть богато белками, углеводами, жирами, а также содержать витамины, минералы, пищевые волокна и большое количество жидкости. При однообразной диете и недостаточном питании потребление витаминов и минералов с пищей ограничено, поэтому рекомендуется дополнительно принимать поливитаминно-минеральные комплексы. При наличии эрозий во рту, дисфагии и сужении пищевода назначают жидкие комплексы. В их составе особенно важны витамины А, группа В, С, D и Е, из минералов — железо, цинк, селен и кальций: витамин А (ретинола пальмитат), масляный раствор 100 000 МЕ/мл перорально на ночь по 10–30 капель (в зависимости от возраста и веса пациента) в течение 2 месяцев. Курсы терапии можно повторять с интервалом в 3 месяца; витамин С (аскорбиновая кислота) 100 мг перорально после еды 3–4 раза в сутки в течение 2 недель. При большом количестве пузырей и эрозий пациент нуждается в восполнении теряемой жидкости [7, 17].
В настоящее время пренатальная диагностика является лучшим способ профилактики тяжелых наследственных заболеваний. Она возможна только при планировании беременности задолго до ее наступления. Медико-генетическое консультирование семьи проводится с целью оценки риска появления больного ребенка в семье, информирования семьи о риске развития наследственного заболевания, о возможных диагностических и терапевтических методах. Показания для консультирования — предыдущее рождение ребенка с ВБЭ, наличие заболевания у одного из родителей, установленное или подозреваемое заболевание в семье. Генетический анализ крови и кожи больного позволяет уточнить тип и подтип ВБЭ, а также обнаружить мутацию соответствующего гена. При наступлении беременности в сроки 10–12 недель проводят биопсию ворсин хориона, в которых ведется поиск уже известной мутации. Быстрое получение результатов (в течение 3–4 дней после взятия материала) позволяет принять решение о прерывании беременности своевременно.
У пациента с БЭ профилактические меры в отношении появления пузырей на коже и слизистых оболочках включают ограничение возможности травмирования кожи (одежда, диета, особенности ухода, неадгезивные повязки, наружные средства, уход за полостью рта). Таким образом, профилактика развития осложнений, диспансеризация, периодический контроль лабораторных показателей для выявления и контроля анемии, полный осмотр пациентов с целью раннего выявления злокачественных опухолей кожи, своевременное лечение зубов будут способствовать существенному облегчению и улучшению качаства жизни пациентов.
Литература
- Lara-Corrales I., Mellerio J. E., Martinez A. E. et al. Dilated cardiomyopathy in epidermolysis bullosa: a retrospective, multicenter study // Pediatr Dermatol. 2010; 27 (3): p. 238–243.
- Гараева З. Ш., Юсупова Л. А., Мавлютова Г. И., Юнусова Е. И., Мирзина Д. Р. Буллезный эпидермолиз. Сборник материалов Всероссийской научно-практической конференции с международным участием «Казанские дерматологические чтения: синтез науки и практики». Казань, 2016. С. 8–20.
- Коталевская Ю. Ю., Кропачева В. В., Марычева Н. М. Буллезный эпидермолиз — состояние проблемы в России. Материалы I Евразийской Конференции по редким заболеваниям и редким лекарствам и III Всероссийской Конференции по редким заболеваниям и редко применяемым медицинским технологиям «Дорога жизни». М., 2012. С. 15–19.
- Mitsuhashi Y., Hashimoto I. Genetic abnormalities and clinical classification of epidermolysis bullosa // Arch Dermatol Res. 2003; 295 (Suppl 1.): 29–33.
- Uitto J., Richard G. Progress in epidermolysis bullosa: genetic classification and clinical implications // Am J Med Genet C Semin Med Genet. 2004; 131 C (1): 61–74.
- Юсупoвa Л. A. Иммунопатология хронических дерматозов. Казань: НБ КГМА, 2017. 108 с.
- Федеральные клинические рекомендации. Дерматовенерология 2015: Болезни кожи. Инфекции, передаваемые половым путем. 5-е изд., перераб. и доп. М.: Деловой экспресс. 2016. 768 с.
- Иванов О. И. Кожные и венерические болезни: Шико. М., 2006. 133–137 с.
- Альбанова В. И., Чикин В. В., Епишев Р. В. К вопросу о диагностике врожденного буллезного эпидермолиза // Вестник дерматологии и венерологии. 2014. № 3. С. 53–59.
- Pohla-Gubo G., Cepeda-Valdes R., Hintner H. Immunofluorescence mapping for the diagnosis of epidermolysis bullosa // Dermatol Clin. 2010; 28: p. 201–210.
- Cepeda-Valdés R., Pohla-Gubo G., Borbolla-Escoboza J. R. et al. Immunofluorescence mapping for diagnosis of congenital epidermolysis bullosa // Actas Dermosifiliogr. 2010; 101 (8): p. 673–682.
- Intong L. R., Murrell D. F. How to take skin biopsies for epidermolysis bullosa // Dermatol Clin. 2010; 28: p. 197–200.
- Rodeck C. H., Eady R. A., Gosden C. M. Prenatal diagnosis of epidermolysis bullosa letalis // Lancet. 1980; 1: p. 949–952.
- Pasmooij A. M., Pas H. H., Bolling M. C., Jonkman M. F. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3 // J Clin Invest. 2007; 117 (5): p. 1240–1248.
- Pasmooij A. M., Pas H. H., Deviaene F. C. et al. Multiple correcting COL17 A1 mutations in patients with revertant mosaicism of epidermolysis bullosa // Am J Hum Genet. 2005; 77 (5): p. 727–740.
- Renwick P., Ogilvie C. M. Preimplantation genetic diagnosis for monogenic diseases: overview and emerging issues // Expert Rev Mol Diagn. 2007; 7: p. 33–43.
- Boeira V. L., Souza E. S., Rocha Bde O. et al. Inherited epidermolysis bullosa: clinical and therapeutic aspects // An Bras Dermatol 2013; 88 (2): p. 185–198.
Л. А. Юсупова*, 1, доктор медицинских наук, профессор
Е. И. Юнусова*, кандидат медицинских наук
З. Ш. Гараева*, кандидат медицинских наук
Г. И. Мавлютова*, кандидат медицинских наук
М. А. Морозова**
* ГБОУ ДПО КГМА МЗ РФ, Казань
** ГАУЗ РККВД, Казань
1 Контактная информация: [email protected]
Современные особенности клиники, диагностики и терапии больных буллезным эпидермолизом/ Л. А. Юсупова, Е. И. Юнусова, З. Ш. Гараева, Г. И. Мавлютова, М. А. Морозова
Для цитирования: Лечащий врач № 1/2018; Номера страниц в выпуске: 71-74
Теги: заболевания кожи, генетические мутации, белки
Трудности дифференциальной диагностики подтипов пограничного типа буллезного эпидермолиза: описание двух клинических наблюдений | Коталевская
1. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, McGrath JA, Mellerio JE, Murrell DF, Shimizu H, Uitto J, Vahlquist A, Woodley D, Zambruno G. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58(6):931–50. doi: 10.1016/j.jaad.2008.02.004.
2. Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, Marinkovich MP, Martinez AE, McGrath JA, Mellerio JE, Moss C, Murrell DF, Shimizu H, Uitto J, Woodley D, Zambruno G. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–26. doi: 10.1016/j.jaad.2014.01.903.
3. Fine JD, Bauer EA, McGuire J, Moshell A, editors. Epidermolysis bullosa: Clinical, epidemiologic, and laboratory advances and the findings of the National Epidermolysis Bullosa Registry. Baltimore, MD: Johns Hopkins University Press; 1999.
4. Vahlquist A, Tasanen K. Epidermolysis bullosa care in Scandinavia. Dermatol Clin. 2010;28(2): 425–7, xv. doi: 10.1016/j.det.2010.02.018.
5. Horn HM, Priestley GC, Eady RA, Tidman MJ. The prevalence of epidermolysis bullosa in Scotland. Br J Dermatol. 1997;136(4):560–4. doi: 10.1046/j.1365-2133.1997.d01-1235.x.
6. Кубанов АА, Альбанова ВИ, Карамова АЭ, Чикин ВВ, Мелехина ЛЕ, Богданова ЕВ. Распространенность врожденного буллезного эпидермолиза у населения Российской Федерации. Вестник дерматологии и венерологии. 2015;(3):21–30. doi: 10.25208/0042-4609-2015-0-3-21-30.
7. Pfendner EG, Lucky AW. Junctional epidermolysis bullosa. GeneReviews [Internet]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1125/.
8. Kelly-Mancuso G, Kopelan B, Azizkhan RG, Lucky AW. Junctional epidermolysis bullosa incidence and survival: 5-year experience of the Dystrophic Epidermolysis Bullosa Research Association of America (DebRA) nurse educator, 2007 to 2011. Pediatr Dermatol. 2014;31(2): 159–62. doi: 10.1111/pde.12157.
9. Файн Дж.-Д., Хинтнер Х, ред. Буллезный эпидермолиз. Пер. с англ. под ред. Ю.Ю. Коталевской. М.: Практика; 2014. 358 с.
10. Kowalewski C, Bremer J, Gostynski A, Wertheim-Tysarowska K, Wozniak K, Bal J, Jonkman MF, Pasmooij AM. Amelioration of junctional epidermolysis bullosa due to exon skipping. Br J Dermatol. 2016;174(6):1375–9. doi: 10.1111/bjd.14374.
11. Barzegar M, Mozafari N, Kariminejad A, Asadikani Z, Ozoemena L, McGrath JA. A new homozygous nonsense mutation in LAMA3A underlying laryngo-onycho-cutaneous syndrome. Br J Dermatol. 2013;169(6):1353–6. doi: 10.1111/bjd.12522.
12. Salvestrini C, McGrath JA, Ozoemena L, Husain K, Buhamrah E, Sabery N, Leichtner A, Rufo PA, Perez-Atayde A, Orteu CH, Torrente F, Heuschkel RB, Thomson MA, Murch SH. Desquamative enteropathy and pyloric atresia without skin disease caused by a novel intracellular beta4 integrin mutation. J Pediatr Gastroenterol Nutr. 2008;47(5):585–91. doi: 10.1097/MPG.0b013e31817af98d.
13. Fine JD. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016;152(11):1231–8. doi: 10.1001/jamadermatol.2016.2473.
14. Shabbir G, Hassan M, Kazmi A. Laryngo-onycho-cutaneous syndrome – a study of 22 cases. Biomedica. 1986;2:15–25.
15. ЛОК (ларинго-онихо-кутанный) синдром. ORPHA:2407 [Интернет]. Доступно на: https://www.orpha.net/data/patho/RU/LOC-syndrome-RUrusAbs2233.pdf.
16. Kadyan A, Aralikatti A, Shah S, Jewell R, Paul L, Darling J, Wood M, Gooi J, Morrell AJ, Newton Bishop JA, Marr JE. Laryngo-onycho-cutaneous syndrome. Ophthalmology. 2010;117(5):1056– 1056.e2. doi: 10.1016/j.ophtha.2009.11.019.
17. Cohn HI, Murrell DF. Laryngo-onycho-cutaneous syndrome. Dermatol Clin. 2010;28(1): 89–92. doi: 10.1016/j.det.2009.10.010.
18. Hirsch T, Rothoeft T, Teig N, Bauer JW, Pellegrini G, De Rosa L, Scaglione D, Reichelt J, Klausegger A, Kneisz D, Romano O, Secone Seconetti A, Contin R, Enzo E, Jurman I, Carulli S, Jacobsen F, Luecke T, Lehnhardt M, Fischer M, Kueckelhaus M, Quaglino D, Morgante M, Bicciato S, Bondanza S, De Luca M. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551(7680): 327–32. doi: 10.1038/nature24487.
19. Fine JD, Johnson LB, Weiner M, Suchindran C. Cause-specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr. 2008;152(2):276–80. doi: 10.1016/j.jpeds.2007.06.039.
20. Fine JD, Johnson LB, Weiner M, Stein A, Cash S, Deleoz J, Devries DT, Suchindran C. Eye involvement in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry. Am J Ophthalmol. 2004;138(2):254– 62. doi: 10.1016/j.ajo.2004.03.034.
21. Pearson RW, Potter B, Strauss F. Epidermolysis bullosa hereditaria letalis. Clinical and histological manifestations and course of the disease. Arch Dermatol. 1974;109(3):349–55. doi: 10.1001/archderm.1974.01630030009001.
22. Yuen WY, Duipmans JC, Molenbuur B, Herpertz I, Mandema JM, Jonkman MF. Longterm follow-up of patients with Herlitz-type junctional epidermolysis bullosa. Br J Dermatol. 2012;167(2):374–82. doi: 10.1111/j.1365-2133.2012.10997.x.
23. Vidal F, Baudoin C, Miquel C, Galliano MF, Christiano AM, Uitto J, Ortonne JP, Meneguzzi G. Cloning of the laminin alpha 3 chain gene (LAMA3) and identification of a homozygous deletion in a patient with Herlitz junctional epidermolysis bullosa. Genomics. 1995;30(2):273–80. doi: 10.1006/geno.1995.9877.
24. Schneider H, Mühle C, Pacho F. Biological function of laminin-5 and pathogenic impact of its deficiency. Eur J Cell Biol. 2007;86(11–12): 701–17. doi: 10.1016/j.ejcb.2006.07.004.
25. Hammersen J, Has C, Naumann-Bartsch N, Stachel D, Kiritsi D, Söder S, Tardieu M, Metzler M, Bruckner-Tuderman L, Schneider H. Genotype, Clinical Course, and Therapeutic Decision Making in 76 Infants with Severe Generalized Junctional Epidermolysis Bullosa. J Invest Dermatol. 2016;136(11):2150–7. doi: 10.1016/j.jid.2016.06.609.
26. Pacho F, Zambruno G, Calabresi V, Kiritsi D, Schneider H. Efficiency of translation termination in humans is highly dependent upon nucleotides in the neighbourhood of a (premature) termination codon. J Med Genet. 2011;48(9):640–4. doi: 10.1136/jmg.2011.089615.
27. Mühle C, Jiang QJ, Charlesworth A, Bruckner-Tuderman L, Meneguzzi G, Schneider H. Novel and recurrent mutations in the laminin-5 genes causing lethal junctional epidermolysis bullosa: molecular basis and clinical course of Herlitz disease. Hum Genet. 2005;116(1–2):33–42. doi: 10.1007/s00439-004-1210-y.
28. Ayoub N, Tomb R, Charlesworth A, Meneguzzi G. Junctional epidermolysis bullosa. Identification of a new mutation in two Lebanese families. Ann Dermatol Venereol. 2005;132(6–7 Pt 1):550–3.
29. Laryngoonychocutaneous syndrome; LOCS. OMIM # 245660 [Internet]. Available from: http://omim.org/entry/245660#1.
30. McLean WH, Irvine AD, Hamill KJ, Whittock NV, Coleman-Campbell CM, Mellerio JE, Ashton GS, Dopping-Hepenstal PJ, Eady RA, Jamil T, Phillips R, Shabbir SG, Haroon TS, Khurshid K, Moore JE, Page B, Darling J, Atherton DJ, Van Steensel MA, Munro CS, Smith FJ, McGrath JA. An unusual N-terminal deletion of the laminin alpha3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome. Hum Mol Genet. 2003;12(18):2395–409. doi: 10.1093/hmg/ddg234.
31. Prodinger C, Klausegger A, Diem A, Bauer JW, Laimer M. Laryngo-onycho-cutaneous (-like) syndrome due to mutated Plectin. J Eur Acad Dermatol Venereol. 2017;31(8):e373–4. doi: 10.1111/jdv.14173.
Буллезный эпидермолиз
Буллезный эпидермолиз (БЭ) является редким генетическим заболеванием, которое поражает кожу и глубокие слизистые оболочки. Из-за наследственных факторов, кожа образует пузыри и эрозии и имеет тенденцию к сдиранию с сильной болью в результате незначительной травмы или трения, или даже спонтанно. По этой причине детей с этим редким заболеванием называют «дети-бабочки», потому что их кожа хрупкая, как крылья бабочек. Жизнь с БЭ сравнима с жизнью с третьей степенью ожога. Это болезненное состояние требует принятие родителями специальных мер предосторожности в обращении и питании ребенка. Страдающему ежедневно, порой по несколько часов, накладывают бинты для защиты ран, часто требуется антибиотикотерапия, чтобы противостоять возникновению инфекции. В более серьезных случаях может потребоваться хирургическая дилатация пищевода и реконструкция рук, есть также риск развития рака кожи и осложнений на почки. БЭ имеет также большое влияние на психологическую и общественную жизнь, и не только потому, что это «видимая» болезнь, которая вызывает любопытство других, но и потому, что вызывает затруднения, а иногда и делает невозможным такие естественные жесты, как бег, игры, обнимать и быть обнятым.
Существуют три основные формы БЭ (простая, пограничная, дистрофическая), каждая из которых включает в себя другие подвиды и каждый случай может проявляться с симптомами, которые изменяются в широких пределах, от легких до очень тяжелых и даже летальных. Это потому, что это генетическое заболевание, вызванное наследственными факторами, которые могут быть доминирующими (передача от больного родителя) или рецессивными (передача от обоих родителей-носителей) и, следовательно, с участием различных генов.
В 50% случаев БЭ проявляется в виде простой формы, как правило, менее серьезная, в основном с участием рук и ног.
Буллезный эпидермолиз: роль одежды
Выбор одежды имеет решающее значение для людей, страдающих от БЭ, и особого внимания требует особенно нижнее белье, которое находится в непосредственном контакте с кожей и должно быть особенно мягким и ограничивать как можно больше трений.
Лечебная одежда DermaSilk, специально разработанная для тех, кто страдает от заболеваний кожи и слизистых оболочек, оказалась очень полезной и эффективной при буллезном эпидермолизе, наблюдалось уменьшение симптомов и значительное улучшение качества жизни пациентов.
Благодаря своим характеристикам, лечебная одежда DermaSilk из фиброина шелка с постоянной антимикробной защитой способна:
- уменьшить механическое трение, защищая кожу от покраснения и раздражения;
- уменьшить зуд и воспаление;
- контролировать и предотвратить инфекции от бактерий, грибов и дрожжей;
- контролировать потоотделение и поддерживать правильный баланс влаги;
- способствует заживлению травм и регенерации кожи.
Эффективность DermaSilk в уменьшении симптомов и улучшении качества жизни пациентов с БЭ была выделена из исследования, проведенного в EB-Haus Austria – Больница Зальцбурга.
Скачать PDF с исследованием и другими полезными советами об одежде Dermasilk.
По этой причине, после положительных клинических оценок и активного участия DEBRA Italia Onlus (Ассоциация пациентов с БЭ), начиная с 2010 года, в Италии, люди с БЭ, имеют право на возмещение продукции DermaSilk Национальной службой здравоохранения.
В России благотворительный фонд «Б.Э.Л.А Дети-бабочки», который занимается всесторонней помощью детям с редким генетическим заболеванием – буллёзным эпидермолизом (БЭ), упоминает белье Dermasilk и постельное белье Microair Pristine как не медикаментозные меры поведенческой терапии при БЭ. Подробнее …
Буллезный эпидермолиз — NORD (Национальная организация по редким заболеваниям)
ТЕКСТОВЫЕ КНИГИ
Fine JD, Hintner H., eds. Жизнь с буллезным эпидермолизом (БЭ): этиология, диагностика, многопрофильная помощь и терапия, Нью-Йорк: Springer Wien; 2009.
Fine JD, Bauer E, McGuire J, Moshell A, eds. Буллезный эпидермолиз: клинические, эпидемиологические и лабораторные достижения и результаты Национального реестра буллезного эпидермолиза 1-е издание The Johns Hopkins University Press; 1999 г.
Weston WL, Lane AT, Morelli JG, ред. Цветной учебник детской дерматологии, 4-е издание Mosby / Elsevier, 2007: 348-354.
ОБЗОР СТАТЬИ
Мюррелл Д.Ф. (ред.) (2010): Буллезный эпидермолиз: Часть I — Патогенез и клинические особенности. Дерматологические клиники, Том 28/1. Филадельфия: W.B. Saunders Elsevier.
Мюррелл Д.Ф. (ред.) (2010): Буллезный эпидермолиз: Часть II — Диагностика и лечение. Дерматологические клиники, Том 28/2. Филадельфия: W.B. Saunders Elsevier.
Fine JD, Mellerio JE. Внекожные проявления и осложнения наследственного буллезного эпидермолиза: часть I. Ткани, ассоциированные с эпителием.
J Am Acad Dermatol. 2009 сентябрь; 61 (3): 367-84.
Fine JD, Mellerio JE. Внекожные проявления и осложнения наследственного буллезного эпидермолиза: часть II. Другие органы. J Am Acad Dermatol. 2009 сентябрь; 61 (3): 387-402.
Pfendne EG, Bruckner A, Conget P, Mellerio J, Palisson F, Lucky AW. Фундаментальная наука о буллезном эпидермолизе, его диагностическая и молекулярная характеристика: Материалы II Международного симпозиума по буллезному эпидермолизу.Сантьяго, Чили, 2005 Международный журнал дерматологии. 2007; 46 (8): 781-794.
Азизхан Р.Г., Дениер Дж.Э., Меллерио Дж.Э., Гонсалес Р., Бачигалупо М., Кантор А., Пассалаква Г., Палиссон Ф., Лаки А.В. Хирургическое лечение буллезного эпидермолиза: материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 г. Международный журнал дерматологии. 2007: 46 (8): 801-808.
Mellerio JE, Weiner M, Denyer JE, Pillay EI, Lucky AW, Bruckner A, Palisson F. Медицинское лечение буллезного эпидермолиза: Материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 Международный журнал дерматологии 2007; 46 (8): 795-800.
Lucky AW, Pfendner E, Pillay E, Paskel J, Weiner M, Palisson F. Психосоциальные аспекты буллезного эпидермолиза: Материалы II Международного симпозиума по буллезному эпидермолизу, Сантьяго, Чили, 2005 Международный журнал дерматологии. 2007; 46 (8): 809-814.
ИНТЕРНЕТ
Debra International. Уход за больным БЭ. Доступно по адресу: http://www.debra-international.org/patients/caring-for-someone-with-eb.html По состоянию на 30 мая 2013 г.
Маринкович М.П.Буллезный эпидермолиз Обновлено: 10 августа 2012 г. Medscape. Доступно по адресу: http://emedicine.medscape.com/article/1062939-overview По состоянию на 30 мая 2013 г.
Пфенднер Э., Брукнер А. Простой буллезный эпидермолиз. Последнее обновление 1 сентября 2011 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013 гг. Доступно на http://www.genetests.org. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Дистрофический буллезный эпидермолиз.Последнее обновление 4 ноября 2010 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013 гг. Доступно на http://www.genetests.org. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Буллезный узловой эпидермолиз. Размещено: 2 февраля 2008 г. В: Обзоры генов на сайте GeneTests: информационный ресурс по медицинской генетике (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013 гг. Доступно по адресу http: //www.genetests.орг. По состоянию на 30 мая 2013 г.
Pfendner E, Lucky AW. Буллезный эпидермолиз с атрезией привратника. Последнее обновление: 14 февраля 2013 г. В: GeneReviews at GeneTests: Medical Genetics Information Resource (онлайн-база данных). Авторские права, Вашингтонский университет, Сиэтл. 1997-2013 гг. Доступно по адресу http://www.genetests.org По состоянию на 30 мая 2013 г.
Буллезный эпидермолиз — NHS
Буллезный эпидермолиз (БЭ) — это название группы редких наследственных кожных заболеваний, при которых кожа становится очень хрупкой.Любая травма или трение кожи могут вызвать болезненные волдыри.
Информация:
Консультации по коронавирусу
Получите консультацию по коронавирусу и буллезному эпидермолизу (БЭ) от DEBRA
Симптомы буллезного эпидермолиза
Основные симптомы всех типов БЭ включают:
- кожа, которая легко покрывается волдырями
- волдыри во рту
- волдыри на руках и подошвах ног
- кожа с рубцами, иногда с небольшими белыми пятна, называемые милиами
- утолщение кожи и ногтей
Кредит:
НАУЧНАЯ ФОТОБИБЛИОТЕКА
https: // www.sciencephoto.com/media/151100/view
Типы буллезного эпидермолиза
Три основных типа БЭ:
- простой буллезный эпидермолиз (EBS) — наиболее распространенный тип, который имеет тенденцию быть более легким с низким риском серьезных осложнений
- дистрофический буллезный эпидермолиз (DEB) — от легкой до тяжелой
- Буллезный узловой эпидермолиз (JEB) — самый редкий и самый тяжелый тип
Тип отражает, где на теле появляются волдыри и какой слой кожи поражен .
Существует также множество вариантов этих трех основных типов БЭ, каждый из которых имеет несколько разные симптомы.
Подробнее о симптомах различных типов буллезного эпидермолиза.
Диагностика EB
EB обычно диагностируется у младенцев и детей вашей неонатальной бригадой, поскольку симптомы часто очевидны с рождения. Но некоторые более легкие типы БЭ могут быть диагностированы только в зрелом возрасте.
Если есть подозрение, что у вашего ребенка такое заболевание, его направят к кожному специалисту (дерматологу).
Специалист проведет обследование для определения типа БЭ и поможет составить план лечения. Они могут взять небольшой образец кожи (биопсия) для отправки на исследование.
Пренатальное тестирование
В некоторых случаях можно проверить еще не родившегося ребенка на БЭ примерно на 11 неделе беременности.
Это может быть предложено, если известно, что вы или ваш партнер являетесь носителем дефектного гена, связанного с БЭ, и есть риск рождения ребенка с тяжелым типом БЭ.
Если тест подтвердит, что у вашего ребенка БЭ, вам будут предложены консультации и рекомендации, которые помогут вам принять обоснованное решение о том, как вы хотите продолжить беременность.
Пренатальные тесты включают амниоцентез и забор проб ворсин хориона.
Причины буллезного эпидермолиза
EB вызывается дефектным геном (генная мутация), который делает кожу более хрупкой.
Обычно ребенок с EB унаследует дефектный ген от родителя, у которого также есть EB.
Также возможно, что ребенок с EB унаследовал дефектный ген от обоих родителей, которые являются просто «носителями», но сами не имеют EB.
Лечение буллезного эпидермолиза
В настоящее время лекарства от БЭ не существует, поэтому лечение направлено на облегчение симптомов и предотвращение развития осложнений, таких как инфекция.
Группа медицинских специалистов поможет вам решить, какое лечение лучше всего подходит для вашего ребенка, и посоветует, как жить с этим заболеванием.
Вы можете лечить БЭ в домашних условиях:
- вскрывать волдыри стерильной иглой
- накладывать защитные повязки
- избегать вещей, ухудшающих состояние
Лекарства можно использовать для лечения инфекции или уменьшения боли. Операция может потребоваться, если БЭ вызывает сужение пищевода или проблемы с руками.
Подробнее о лечении буллезного эпидермолиза.
Приобретенный буллезный эпидермолиз
Приобретенный буллезный эпидермолиз (ББЭ) — приобретенная форма БЭ с аналогичными симптомами.
Как и EB, EBA легко вызывает образование волдырей. Это также может повлиять на ротовую полость, горло и пищеварительный тракт.
Но EBA не передается по наследству, и симптомы обычно не проявляются до более позднего возраста.
Это аутоиммунное заболевание, которое означает, что ваша иммунная система начинает атаковать здоровые ткани тела. Точно не известно, что вызывает это.
EBA — очень редкое заболевание, которое чаще встречается у людей старше 40 лет.
Благотворительные организации и группы поддержки
Если вашему ребенку поставили диагноз БЭ, это может быть пугающим и подавляющим опытом.Вероятно, вы захотите узнать как можно больше о состоянии и доступных методах лечения.
DEBRA — это национальная благотворительная организация, которая предоставляет помощь, советы и поддержку людям в Великобритании, живущим с EB.
DEBRA International — это всемирная сеть национальных групп, работающих от имени людей, пострадавших от БЭ.
Поддержка лиц, осуществляющих уход
При уходе за ребенком со сложным и тяжелым заболеванием, таким как БЭ, важно не пренебрегать своим здоровьем и благополучием.
Прочтите о перерывах в уходе и временном уходе и получите советы по уходу за ребенком-инвалидом.
Информация о вас
Если у вас или вашего ребенка есть EB, ваша клиническая бригада передаст информацию о вас или вашем ребенке в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния. Вы можете отказаться от регистрации в любое время.
Подробнее о реестре
Последняя проверка страницы: 19 марта 2018 г.
Срок следующей проверки: 19 марта 2021 г.
Простой буллезный эпидермолиз: MedlinePlus Genetics
Простой буллезный эпидермолиз — это одно из группы генетических состояний, называемых буллезным эпидермолизом, при которых кожа становится очень хрупкой и легко образуется волдыря. Волдыри и участки потери кожи (эрозии) возникают в результате незначительной травмы или трения, например, трения или царапин.Простой буллезный эпидермолиз — одна из основных форм буллезного эпидермолиза. Признаки и симптомы этого состояния у разных людей сильно различаются. В легких случаях образование волдырей поражает, прежде всего, руки и ноги, и волдыри обычно заживают, не оставляя шрамов. Тяжелые случаи этого состояния включают широко распространенные волдыри, которые могут привести к инфекциям, обезвоживанию и другим проблемам со здоровьем. Тяжелые случаи могут быть опасными для жизни в младенчестве.
Исследователи выделили четыре основных типа простого буллезного эпидермолиза.
Хотя типы различаются по степени серьезности, их особенности значительно перекрываются, и они вызваны мутациями в одних и тех же генах. Большинство исследователей теперь рассматривают основные формы этого состояния как часть одного расстройства с рядом признаков и симптомов.
Самая легкая форма простого буллезного эпидермолиза, известная как локализованный тип (ранее называвшаяся типом Вебера-Кокейна), характеризуется образованием волдырей на коже, которое начинается в любое время между детством и взрослостью и обычно ограничивается руками и ногами.В более зрелом возрасте кожа на ладонях рук и подошвах ног может утолщаться и затвердевать (гиперкератоз).
Тип Даулинга-Меара представляет собой наиболее тяжелую форму простого буллезного эпидермолиза. Обширные и сильные волдыри могут возникать на любом участке тела, включая внутреннюю часть рта, а волдыри могут появляться группами. Волдыри присутствует с рождения и имеет тенденцию к уменьшению с возрастом. У больных также наблюдается аномальный рост ногтей и гиперкератоз ладоней и подошв.
Другая форма простого буллезного эпидермолиза, известная как другой генерализованный тип (ранее называвшаяся типом Кебнера), связана с широко распространенными волдырями, которые появляются при рождении или в раннем младенчестве. Волдыри, как правило, менее серьезны, чем при типе Даулинга-Меары.
Простой буллезный эпидермолиз с пятнистой пигментацией характеризуется участками более темной кожи на туловище, руках и ногах, которые исчезают в зрелом возрасте. Эта форма заболевания также включает образование волдырей на коже в раннем младенчестве, гиперкератоз ладоней и подошв, а также аномальный рост ногтей.
В дополнение к четырем основным типам, описанным выше, исследователи определили еще одно состояние кожи, связанное с простым буллезным эпидермолизом, которое они называют типом Огна. Это вызвано мутациями в гене, который не связан с другими типами простого буллезного эпидермолиза. Неясно, является ли тип Ogna подтипом простого буллезного эпидермолиза или представляет собой отдельную форму буллезного эпидермолиза.
Было предложено несколько других вариантов простого буллезного эпидермолиза, но они оказались очень редкими.
Признаки, симптомы, причины, диагностика, лечение
Обзор
Что такое буллезный эпидермолиз (БЭ)?
Буллезный эпидермолиз (БЭ) — это группа заболеваний соединительной ткани, при которых кожа становится хрупкой, легко покрывается волдырями и рвется. Волдыри и язвы возникают, когда одежда трется о кожу или когда она ударяется.В легких случаях болезнь обычно вызывает появление болезненных волдырей на руках, локтях, коленях и ступнях. Однако слезы и волдыри могут появиться на любом участке тела.
В некоторых случаях волдыри образуются внутри тела в таких местах, как рот, пищевод, другие внутренние органы или глаза. Когда волдыри заживают, они могут вызвать болезненные рубцы. В тяжелых случаях волдыри и шрамы могут повредить внутренние органы и ткани настолько, что приведет к летальному исходу.
Кто заболевает буллезным эпидермолизом (БЭ)?
Примерно 1 из 50 000 жителей США.С. у ЭБ. Он одинаково поражает мужчин и женщин и диагностируется у людей всех рас и этнических групп. Риск заболеть БЭ выше у людей, у которых есть родители с этим заболеванием.
Симптомы и причины
Каковы признаки и симптомы буллезного эпидермолиза (БЭ)?
Врачи классифицируют буллезный эпидермолиз (БЭ) на четыре основных типа, каждый из которых имеет множество подтипов в зависимости от задействованных генов и места появления волдырей.Четыре типа EB:
- Симплекс: Волдыри появляются на верхнем слое кожи и редко появляются рубцы
- Узловой: Волдыри образуются во рту и дыхательных путях
- Дистрофия: Волдыри растут в дерме (нижний слой кожи)
- Синдром Киндлера: Волдыри могут образовываться во всех слоях кожи
Признаки и симптомы БЭ зависят от типа и обычно появляются, когда больной является младенцем или малышом.Некоторые симптомы совпадают между типами. Симптомы БЭ включают:
- Волдыри на коже (кистях, стопах, локтях и коленях) или внутри тела
- Утолщенные мозоли на ладонях и подошвах
- Анемия (низкий уровень эритроцитов)
- Сросшиеся (прикрепленные) пальцы рук или ног
- Деформированные и / или утолщенные ногти на руках и ногах
- Маленькие белые бугорки на коже — милиа
- Затруднение глотания (дисфагия)
- Отсутствие нормального роста у младенца
Что вызывает буллезный эпидермолиз (БЭ)?
Мутация (дефект) в одном из 18 генов вызывает БЭ.У людей с этим заболеванием отсутствует или поврежден ген, который влияет на белок, используемый для выработки коллагена. Коллаген придает соединительной ткани, такой как кожа, прочность и структуру. Из-за этого дефекта слои кожи (эпидермис и дерма) не связываются друг с другом, как обычно. Это приводит к тому, что кожа становится хрупкой, легко покрывается волдырями и слезами.
EB может передаваться по наследству, что означает, что дефектный ген может передаваться от членов семьи.
В редких случаях EB также может быть аутоиммунным заболеванием, когда иммунная система организма атакует собственные клетки.
Диагностика и тесты
Как врачи диагностируют буллезный эпидермолиз (БЭ)?
Врачи диагностируют буллезный эпидермолиз (БЭ) с помощью теста, называемого биопсией кожи. В этом тесте врач удаляет небольшой образец кожи и изучает его под микроскопом. Генетический тест может подтвердить тип БЭ, идентифицируя дефектный ген.
Ведение и лечение
Какие методы лечения буллезного эпидермолиза (БЭ)?
Нет лекарства от буллезного эпидермолиза (БЭ). Лечение направлено на предотвращение образования волдырей, уход за волдырями и кожей, чтобы не возникало осложнений, лечение проблем с питанием, которые могут возникнуть из-за волдырей во рту или пищеводе, и облегчение боли.
Во избежание повреждений и трения, которые могут вызвать образование волдырей или разрывов на коже, врачи рекомендуют:
- Носить мягкую свободную одежду.
- Избегать перегрева; поддерживайте в комнатах комфортную и равномерную температуру.
- Держитесь подальше от солнца или пользуйтесь солнцезащитным кремом.
- Наложение специальных повязок для защиты кожи — использовать неклейкие (не прилипающие к коже) повязки и скотч и свернутую марлю.
№
Для лечения волдырей ваш врач может порекомендовать:
- Ежедневное лечение ран мазями.
- Использование лечебных повязок для заживления волдырей и предотвращения инфекции.
- Прием обезболивающих.
Для лечения инфекций ваш врач может порекомендовать:
- Прием антибиотиков внутрь или нанесение крема с антибиотиком.
- Использование специального покрытия для незаживающих ран.
Чтобы предотвратить проблемы с питанием из-за затруднений с приемом пищи из-за волдырей во рту или пищеводе, ваш врач может порекомендовать:
- Использование детской бутылочки со специальной соской.
- Кормление ребенка пипеткой или шприцем.
- Добавление жидкости в пюре для разжижения и облегчения употребления в пищу.
- Обеспечение диеты мягкой пищей, такой как супы, пюре, пудинг, яблочное пюре.
- Подача продуктов с теплой (не горячей) температурой.
- Обращение к диетологу для наблюдения за особыми потребностями в питании людей с этим заболеванием.
В тяжелых случаях БЭ может потребоваться хирургическое вмешательство. Может быть проведена операция по расширению пищевода (трубки, ведущей от рта к желудку), который был сужен из-за волдырей и рубцов.Введение питательного зонда непосредственно в желудок, полностью минуя пищевод, — еще один вариант для некоторых людей. Также проводится операция, чтобы отделить сросшиеся пальцы рук и ног от волдырей.
Каковы осложнения или побочные эффекты буллезного эпидермолиза (БЭ)?
Тяжелые случаи БЭ могут привести к потере зрения (при появлении волдырей в глазу). Серьезные обезображивающие шрамы и деформации кожи / мышц, затрудняющие движение пальцев, кистей, стоп и суставов.Некоторые люди с БЭ имеют повышенный риск развития рака кожи, называемого плоскоклеточной карциномой. Иногда смерть может наступить в младенчестве из-за тяжелой инфекции (сепсиса), проблем с дыханием из-за закупорки дыхательных путей, обезвоживания и недоедания.
Профилактика
Можно ли предотвратить буллезный эпидермолиз (БЭ)?
Поскольку он является генетическим, невозможно предотвратить буллезный эпидермолиз (БЭ).Люди с семейным анамнезом БЭ, которые думают о том, чтобы стать родителями, могут использовать генетические тесты, чтобы принять решение о том, как вырастить свою семью.
Перспективы / Прогноз
Каковы перспективы людей с буллезным эпидермолизом (БЭ)?
Перспективы людей с буллезным эпидермолизом (БЭ) зависят от типа и степени тяжести.Тяжелые формы заболевания могут привести к сильной боли, обезображиванию, инвалидности, незаживающим ранам и ранней смерти.
Врачи могут помочь вам справиться с симптомами с помощью надлежащего лечения и, при необходимости, лекарств от боли. Профилактические меры — обширное обертывание кожи через день неклейкими защитными повязками — и регулярное купание и уход за раной могут помочь справиться с воздействием БЭ.
Ресурсы
Какая поддержка предоставляется людям с буллезным эпидермолизом (БЭ)?
Существует множество ресурсов для поддержки людей с БЭ и лиц, ухаживающих за ними.Вы можете присоединиться к группе поддержки, чтобы поделиться опытом и способами борьбы с болезнью.
Американская ассоциация по исследованию буллезного дистрофического эпидермолиза (DEBRA) и Партнерство по исследованиям EB — два наиболее распространенных ресурса для людей, которым нужна дополнительная информация и поддержка:
Буллезный эпидермолиз: эффекты, типы и симптомы
Буллезный эпидермолиз — это редкая группа наследственных заболеваний, вызывающих хрупкую кожу, которая легко образует волдыри.Волдыри часто появляются в результате царапин или трения кожи, контакта с теплом, трением или незначительной травмы.
Буллезный эпидермолиз (БЭ) вызывается дефектом или мутацией в гене кератина или коллагена и влияет на соединительные ткани.
Могут возникать три основных типа БЭ, и предыдущие исследования показывают, что 6,5 на каждый миллион новорожденных в США могут иметь один из этих типов.
Риск одинаков, независимо от пола и этнической принадлежности.
У людей с БЭ очень хрупкая кожа. Даже очень легкое трение может вызвать появление волдырей, поскольку слои кожи перемещаются независимо и разделяются.
Лекарства от БЭ в настоящее время не существует. Лечение направлено на облегчение симптомов боли, риска инфицирования и осложнений.
Здоровая кожа человека состоит из двух слоев:
- эпидермиса, который является внешней частью,
- дермы, которая является внутренней частью
Обычно между слоями существуют белковые якоря.Эти якоря состоят из коллагена и предотвращают срезание или перемещение двух слоев кожи независимо друг от друга.
Люди с БЭ не имеют этих протеиновых якорей, поэтому при трении кожи два слоя трутся друг о друга и разделяются. В результате появляются болезненные язвы и волдыри.
Волдыри могут также возникать на слизистых оболочках внутри тела, например, во рту и пищеводе. Иногда это делает практически невозможным употребление в пищу твердых веществ.БЭ также может поражать мочевыводящие пути и мочевой пузырь, вызывая болезненные ощущения при мочеиспускании.
Врачи часто называют молодых людей с БЭ «детьми-бабочками», потому что их кожа хрупкая, как крылья бабочки.
EB может иметь степень тяжести от легкой до опасной для жизни. При легкой форме БЭ волдыри появляются вокруг кистей и стоп. Тяжелая форма БЭ часто поражает все тело, и некоторые осложнения, такие как инфекция, трудности с кормлением и потеря питательных веществ через кожу, могут быть фатальными.
При БЭ раны заживают очень медленно. Это может вызвать значительные рубцы, физическую деформацию и инвалидность. Люди с тяжелыми формами БЭ имеют значительно более высокий риск развития рака кожи.
Встречаются три основных типа БЭ:
- Наиболее распространенным типом БЭ является простой буллезный эпидермолиз (EBS). На внешнем слое кожи образуются волдыри.
- При дистрофическом буллезном эпидермолизе (ДЭБ) пузыри образуются как на внешнем, так и на внутреннем слоях кожи.
- Буллезный узловой эпидермолиз (ЯБЭ) — наиболее тяжелая форма. Однако это тоже редкость. Волдыри образуются на стыке внешнего и внутреннего слоев кожи.
У каждого типа есть несколько подтипов. На сегодняшний день медицинские эксперты выявили более 30 подтипов БЭ.
У человека с БЭ очень хрупкая кожа, которая повреждается при малейшем прикосновении. Волдыри могут возникнуть даже при легком растирании, ударе или ударе. Одежда, которая соприкасается с кожей или трется о нее, также может увеличить риск образования пузырей.
При очень легких проявлениях симптомы могут развиться только в более позднем возрасте. Когда появляются волдыри, они могут зажить с минимальным рубцеванием или без него.
Конкретные признаки и симптомы зависят от типа БЭ у человека. Обычно они включают:
- волдыри на коже, волосистой части головы, вокруг глаз и носа
- разрыв кожи
- кожа, которая выглядит очень тонкой
- кожа, которая отпадает
- алопеция или выпадение волос
- мили, или очень маленькие белые бугорки на коже
- потеря ногтей на руках, ногах или обоих ногтях, или деформация ногтей
- волдыри или эрозии глаза
- чрезмерное потоотделение
Если БЭ возникает на слизистых оболочках, это может причина:
- трудности с глотанием, если появляются волдыри вокруг рта и горла
- хриплый голос из-за пузырей в горле
- проблемы с дыханием из-за пузырей в верхних дыхательных путях
- болезненное мочеиспускание, которое возникает из-за пузырей в горле мочевыводящие пути
Симптомы обычно развиваются очень рано, часто вскоре после рождения.При синдроме Киндлера, редком подтипе БЭ, волдыри образуются при рождении.
Национальные институты артрита и костно-мышечные и кожные заболевания (NIAMS) предлагают следующие меры для защиты кожи и предотвратить образование пузырей на коже у младенцев, детей и взрослых.
- носить одежду, которая не натирает и не раздражает, например, одежду из чистого хлопка
- с использованием смазки на коже для уменьшения трения
- поддержание комнатной температуры для предотвращения перегрева
- покрытие твердых поверхностей, таких как автомобильные сиденья, с овчиной
- носить варежки во время сна, чтобы предотвратить повреждение, вызванное царапинами
Обнимать всех младенцев и маленьких детей очень важно, и следование этим рекомендациям сделает обнимание ребенка с БЭ безопасным, помогая защитить кожу ребенка от повреждать.Чтобы защитить ребенка от дальнейшего дискомфорта, поднимите его, поместив одну руку под ягодицы ребенка, а другую — за его спину.
Человек с EBS может предотвратить образование волдырей на ногах, избегая длительных прогулок. Те, у кого есть DEB или JEB, должны быть предельно осторожны, чтобы не поцарапать и не поцарапать кожу.
Избегайте острой пищи и твердых, острых продуктов, которые могут порезаться и поцарапаться, защитить волдыри или уменьшить боль во рту.
Убедитесь, что очки не вызывают образование волдырей вокруг носа и ушей.
Ошибочные гены вызывают EB. Человек может унаследовать их от одного или обоих родителей.
Для передачи JEB оба родителя должны иметь дефектный ген. Для всех остальных типов он должен быть только у одного родителя.
В других случаях неисправность может возникнуть во время формирования яйцеклетки или сперматозоидов.
Большинство экспертов считают, что мутация происходит в гене кератина или коллагена.
Врачи диагностируют БЭ у большинства людей в младенчестве. Однако иногда, когда симптомы легкие, врачи ставят диагноз только на более позднем этапе жизни человека.
Врачи могут проводить множество других диагностических тестов на БЭ, в том числе:
- Биопсию, при которой врач берет небольшой образец кожной ткани для анализа. Это может показать, где отделяется кожа и какой у человека БЭ.
- Микроскоп и отраженный свет, который может показать, отсутствуют ли какие-либо белки, необходимые для образования соединительных тканей.
- Мощный электронный микроскоп, позволяющий выявлять структурные дефекты кожи.
- Анализы крови. Врач может проверить образцы крови человека с подозрением на БЭ. Они также могут брать кровь у людей для проверки на генетические проблемы. Это также поможет определить, унаследовал ли человек заболевание от родителя.
- Врач может провести тест на амниоцентез во время беременности. Амниоцентез включает удаление и исследование небольшого количества околоплодных вод, окружающих плод.
- Врач может также рассмотреть возможность взятия пробы ворсинок хориона (CVS), которая включает исследование части мембраны, окружающей плод.
Лекарства от БЭ в настоящее время не существует. Лечение направлено на облегчение симптомов и предотвращение повреждения кожи, инфекций и других осложнений.
Некоторым людям также может потребоваться психологическая и эмоциональная поддержка, чтобы помочь им поддерживать хорошее качество жизни.
- Врач может научить людей, как разбивать большие волдыри безопасно и гигиенично. Умелое прокалывание может позволить жидкости стекать из волдыря, не увеличивая риск заражения или повреждения кожи под ним.
- Врач может назначить антибиотик для местного применения при инфекции.
- Повязки для ран не должны прилипать к коже, поэтому врачи могут порекомендовать определенные типы повязок для облегчения заживления, уменьшения боли и уменьшения вероятности заражения.
- Если поражение не заживает полностью, может потребоваться кожный трансплантат. Прикрытие раны кожей может способствовать процессу заживления.
- Повторяющиеся сильные пузыри и рубцы могут привести к сращению пальцев. Кроме того, мышечная ткань может ненормально укорачиваться, что затрудняет выполнение повседневных задач.В этих ситуациях может потребоваться операция.
- Боль является основным симптомом БЭ, поэтому лечение обычно включает прием обезболивающих.
- Если пузыри в пищеводе затрудняют прием пищи, врач может порекомендовать операцию по расширению пищевода.
- Если человек с БЭ не может глотать пищу, ему может потребоваться гастростомический зонд. Хирург делает отверстие в желудке и пропускает через него зонд для кормления.
У некоторых людей с БЭ могут развиться серьезные осложнения.
- Волдыри могут привести к инфекции и появлению открытых язв.
- Люди с DEB имеют более высокий риск развития плоскоклеточного рака, который является агрессивной формой рака кожи, в возрасте до 35 лет.
- Иногда DEB приводит к слиянию пальцев человека.
- Если БЭ поражает конъюнктиву и другие части глаза, у некоторых людей может наблюдаться потеря зрения.
- Если человек не может легко глотать, у него может развиться недоедание.
- Анемия может развиться из-за хронического воспаления и кровопотери.
Некоторые осложнения БЭ, такие как обезвоживание, инфекция, внутренние волдыри и недоедание, могут быть фатальными, особенно у младенцев.
Q:
Может ли EB развиться в результате других заболеваний или это исключительно генетическое происхождение?
A:
При других условиях преобразование в EB невозможно. Буллезный эпидермолиз — это генетически наследственное заболевание. Иногда один из родителей имеет ген и сам испытывает это заболевание. Вероятность передачи БЭ своему ребенку у этого человека составляет 50 процентов.
В других случаях оба родителя могут нести ген, не подозревая об этом. В таких ситуациях вероятность того, что у ребенка разовьется расстройство, составляет 25 процентов.
Иногда у ребенка может развиться ЭБ, если ни один из родителей не несет ген, но ген в яйцеклетке или сперматозоиде мутирует.
J. Keith Fisher, M.D. Ответы отражают мнение наших медицинских экспертов. Весь контент носит исключительно информационный характер и не может рассматриваться как медицинский совет.
Буллезный эпидермолиз: симптомы, причины, диагностика, лечение
Буллезный эпидермолиз — это редкое генетическое заболевание, которое делает кожу настолько хрупкой, что при малейшем прикосновении она может порваться или покрываться волдырями.Детей, рожденных с ним, часто называют «детьми бабочек», потому что их кожа кажется хрупкой, как крыло бабочки.
Легкие формы со временем могут поправиться. Но тяжелые случаи могут быть болезненными, вызывать другие серьезные проблемы со здоровьем и могут быть опасными для жизни.
Если вы страдаете этим заболеванием, вам необходимо специальное лечение, чтобы ваша нежная кожа оставалась здоровой, насколько это возможно.
Типы
Существует пять основных типов буллезного эпидермолиза. Тип зависит от того, где образуются волдыри.
Простой буллезный эпидермолиз: Самый распространенный тип, впервые обнаруживается у новорожденных. В основном поражаются ладони рук и подошвы ног.
Буллезный соединительный эпидермолиз : Хотя он также впервые появляется у младенцев, это более серьезная форма, вызывающая образование пузырей в глубоких слоях кожи.
Буллезный дистрофический эпидермолиз: Если у вас этот тип кожи, в вашей коже нет коллагена, чтобы удерживать ее вместе, или коллаген, который у вас есть, не работает.Это означает, что слои вашей кожи не срастаются, как должны. Иногда этот тип не проявляется до раннего детства.
Продолжение
Синдром Киндлера: Это смешанное состояние, поскольку волдыри появляются на разных слоях кожи. Он также может вызывать неоднородные изменения цвета кожи под воздействием солнечных лучей.
Приобретенный буллезный эпидермолиз: Эта форма вызывает образование волдырей на руках и ногах, а также на слизистых оболочках, например во рту.
Причины
Практически все типы буллезного эпидермолиза передаются по наследству. Если вы унаследовали определенные генные сбои от родителей, они у вас будут.
Есть одно исключение. Буллезный буллезный эпидермолиз — единственный тип, который не передается по наследству. Это происходит из-за проблемы в вашей иммунной системе.
Симптомы
Обычно признаки буллезного эпидермолиза сначала появляются у младенцев или детей ясельного возраста. Болезненные волдыри на коже — главный симптом. Они могут образовываться где угодно на коже.Иногда они также образуются на глазах или в частях горла, желудка или мочевого пузыря. Если эти волдыри инфицированы или образуют рубцы на коже, они вызывают больше проблем.
Диагноз
Для подтверждения состояния ваш врач может взять небольшой образец кожи и отправить его в лабораторию, где специалисты изучат его под микроскопом.
Лечение
Буллезный эпидермолиз неизлечим. Но есть методы лечения.
Если у вас тяжелый случай, вы будете заботиться о своей коже так же, как человек с ожогом.Вам нужно будет узнать, как проводить ежедневную обработку ран, а также как перевязывать и защищать пораженные участки.
Ваш врач также может назначить лекарство для снятия боли.
В некоторых случаях вам может потребоваться операция. Если у вас есть волдыри, которые срослись между пальцами рук и ног, ваш врач может их разделить. Или, если ваш пищевод, трубка, соединяющая ваш рот с желудком, слишком покрыт рубцами, вы можете сделать операцию по его расширению, чтобы помочь вам есть.
Некоторые люди с буллезным эпидермолизом по-прежнему считают прием пищи слишком болезненным.В этом случае ваш врач может предложить зонд для кормления, чтобы еда попадала прямо в желудок.
Советы по уходу в домашних условиях
Чтобы предотвратить образование волдырей, нужно особенно заботиться о своей коже.
Уменьшить трение. Используйте лосьон, чтобы сохранить кожу влажной и уменьшить трение. Если вы прикрываете раны, используйте только нелипкие повязки, а затем снова неплотно оберните скатанной марлей. Носите свободную одежду без бирок, узких рукавов и швов.
Продолжение
Слить пузырьки. Если их не лечить, они могут заполниться жидкостью и заразиться. Ваш врач может показать вам, как лучше всего их осушить.
Сохраняйте прохладу. Температура воды в ванне должна быть не выше комнатной. Как можно дольше оставайтесь в кондиционировании воздуха, избегайте жары и влажности.
Знайте признаки инфекции. Ваша кожа может покраснеть или стать горячей на ощупь, если она инфицирована. Вы также можете заметить гной или желтые выделения с коркой на этом месте, красную полосу под кожей, или у вас жар или озноб.Если вы заметили какой-либо из этих признаков, немедленно обратитесь к врачу. Вам могут понадобиться антибиотики.
Проверьте свой рацион. Многие люди с буллезным эпидермолизом имеют низкий уровень железа, селена или витамина D. Ваш врач может посоветовать вам обратиться к диетологу по поводу употребления большего количества продуктов, богатых этими витаминами и минералами.
Продолжение
Получите поддержку. Поговорите со своим врачом или с кем-нибудь, кому вы доверяете. Вам также может быть полезно поговорить о своих чувствах с терапевтом или в группе поддержки в вашем районе.
Буллезный эпидермолиз | Детская больница Филадельфии
Буллезный эпидермолиз (БЭ) — редкое генетическое заболевание, вызывающее болезненные волдыри на коже. БЭ может варьироваться от легкой до тяжелой. У некоторых пациентов также появляются волдыри и язвы внутри тела, например, во рту или на слизистой оболочке пищевода (пищевода). Это также может повлиять на другие внутренние органы.
Общим признаком всех людей с БЭ является очень хрупкая кожа. Волдыри могут образовываться в ответ на незначительную травму, даже при трении кожи, и могут прогрессировать, становясь открытыми, кровоточащими, склонными к инфекции и в некоторых случаях рубцами.
Если у вашего ребенка появляются волдыри, особенно без ясной причины, вам следует обратиться к педиатру. Всегда обращайтесь за медицинской помощью, если у вашего ребенка проблемы с глотанием или дыханием, или если у него есть признаки инфекции.
Типы EB
Было идентифицировано несколько типов БЭ. Тип определяется тем, какой слой кожи поражен.
- EB simplex вызывает образование пузырей в нижней части верхнего слоя кожи (эпидермиса).
- Соединительный БЭ вызывает образование пузырей между эпидермисом и дермой (нижний слой кожи)
- Дистрофический БЭ вызывает образование пузырей в верхнем слое нижнего слоя кожи (дермы).
- Синдром Киндлера (затрагивает все слои, описанные выше)
В большинстве случаев буллезный эпидермолиз является наследственным заболеванием. В некоторых формах она передается по наследству от обоих родителей (рецессивное наследование), что означает, что оба родителя должны быть носителями гена, чтобы болезнь появилась у ребенка, даже если сами родители могут не иметь симптомов. При других формах заболевание передается по наследству от одного родителя (доминантное наследование).
Семейный анамнез буллезного эпидермолиза — родители, бабушка или дедушка, тетя или дядя с этим заболеванием — увеличивает вероятность его заражения у ребенка.
Симптомы буллезного эпидермолиза обычно появляются вскоре после рождения или в раннем детстве. Они различаются в зависимости от типа заболевания и могут включать:
- Волдыри на коже, особенно на руках и ногах
- Волдыри во рту
- Волдыри на коже головы при рубцевании и выпадении волос
- Толстые или отсутствующие ногти на руках или ногах или ногти необычной формы
- Утолщение кожи на ладонях и подошвах стоп
- Небольшие белые шишки на коже
- Зудящая, болезненная кожа
- Кожа выглядит необычно тонкой
- Затруднение при глотании (из-за волдырей во рту и горле)
- Запор
- Хриплый плач (признак пузырей и рубцов на голосовых связках)
- Проблемы с зубами, включая кариес, из-за плохо сформированной зубной эмали
- Медленный рост
Симптомы могут не появиться, пока ребенок не начнет хвататься за предметы, ползать или ходить, увеличивая трение в ладонях и подошвах ног.
Осложнения
- Образование пузырей может привести к инфицированию пораженного участка кожи и, если не лечить, кровотока (сепсис)
- Волдыри во рту, горле и пищеварительной системе могут привести к плохому питанию, что, в свою очередь, может снизить способность организма заживлять раны от болезни и поддерживать здоровую скорость роста
- Волдыри и хронические раны на руках и ногах могут привести к сращиванию пальцев рук и ног
- Волдыри в суставах могут привести к ненормальному сгибанию пальцев рук, ног, коленей и локтей
- Подростки и взрослые, страдающие определенными формами заболевания, подвержены более высокому риску развития рака кожи
Когда у ребенка появляются признаки буллезного эпидермолиза, ваш врач спросит, есть ли в семье какие-либо случаи заболевания в анамнезе.Поскольку большинство форм болезни передаются по наследству, любое проявление в большой семье повышает вероятность того, что симптомы ребенка связаны с БЭ.
После осмотра ребенка и сбора семейного анамнеза ваш врач обсудит генетическое тестирование для подтверждения типа БЭ. Анализы могут включать анализ крови или, в некоторых случаях, биопсию.
Из-за характера заболевания и множества систем организма, на которые оно может повлиять, детям с буллезным эпидермолизом может потребоваться специальная поддержка или лечение по целому ряду различных медицинских проблем.
- Предотвращение образования волдырей: Первым лечением ребенка с БЭ является минимизация источников трения на коже, вызывающих образование пузырей. Медицинская бригада вашего ребенка может посоветовать, как держать ребенка, одевать и пеленать его, чтобы предотвратить образование волдырей.
- Уход за раной: Даже при самом лучшем профилактическом уходе образование волдырей неизбежно. Профессиональный уход за раной имеет решающее значение для всех пациентов с этим заболеванием. Могут потребоваться специальные повязки и средства для ухода за ранами, так как контакт со стандартными клеями и повязками может вызвать трение и дополнительное образование пузырей.
- Хроническая боль: Волдыри и язвы могут быть болезненными, поэтому грамотное обезболивание часто является важной частью плана лечения.
- Инфекции: При инфицировании ран могут потребоваться антибиотики.
- Желудочно-кишечные (ЖКТ) проблемы: Пациентам с волдырями и язвами на слизистой оболочке пищеварительной системы может потребоваться лечение хронического запора или диареи, недостаточности питания и витаминов, проблем роста, связанных с плохим усвоением пищи или сужения пищевода. .Лечение может включать изменения в диете, прием витаминов и пищевых добавок, прием лекарств или установку гастростомической трубки (G-трубки).
- Анемия: Пациенты с более тяжелыми случаями БЭ часто страдают хронической анемией из-за дефицита железа, хронических заболеваний и кровопотери из открытых ран. В зависимости от тяжести анемии и причины ее можно лечить витаминами или добавками, диетой, антибиотиками или вливаниями крови.
- Эмоциональные и психологические проблемы: Работа с хроническим заболеванием, таким как буллезный эпидермолиз, может быть стрессовой, а иногда и подавляющей для пациентов и их семей.Психологи могут предложить стратегии преодоления боли и стресса.
- Физическая терапия и трудотерапия: Поскольку активность и диапазон движений ребенка могут быть ограничены, чтобы предотвратить образование волдырей, или из-за боли, связанной с волдырями, часто требуется терапия, чтобы помочь ребенку оставаться физически активным и поддерживать активность в школе и с друзьями.
- Хирургия: В тяжелых случаях может потребоваться операция для устранения проблем, вызванных буллезным эпидермолизом, таких как стриктуры желудочно-кишечного тракта или срастание пальцев рук или ног.
В детской больнице Филадельфии уход за вашим ребенком осуществляется через многопрофильную клинику буллезного эпидермолиза, где они могут получить скоординированное лечение у всех специалистов, которые могут им понадобиться. Наши специалисты имеют опыт лечения детей с БЭ, и у нас есть высококлассные педиатры-узкоспециалисты, которые обеспечат целенаправленное лечение любых симптомов вашего ребенка.
В настоящее время лекарства от БЭ не существует. Пока не найдено лекарство, это считается пожизненным заболеванием.Перспективы для детей с БЭ варьируются в зависимости от типа, который они унаследовали.
В настоящее время проводятся исследования, чтобы лучше понять причины буллезного эпидермолиза и найти новые методы лечения и возможные способы лечения. В настоящее время CHOP участвует в клинических испытаниях БЭ, направленных на поиск новых методов местного лечения, способствующих заживлению ран.
Пожизненная медицинская поддержка необходима детям и взрослым с БЭ. Характер и степень такой помощи будут зависеть от типа и тяжести заболевания, а также от участков тела, пораженных волдырями.
Многопрофильная клиника буллезного эпидермолиза при детской больнице Филадельфии объединяет множество педиатров, необходимых для лечения БЭ вашего ребенка.
Наша команда имеет многолетний опыт оказания специализированной помощи детям с БЭ. Мы тесно сотрудничаем с вами и остальной медицинской бригадой вашего ребенка, чтобы своевременно и эффективно оказывать наилучшую медицинскую помощь. Ваш ребенок может прийти в одну клинику, чтобы увидеть множество разных специалистов, которые могут участвовать в его лечении.
Эти специалисты включают, но не ограничиваются:
- Дерматология
- Гастроэнтерология и питание
- Услуги по обезболиванию
- Психология
- Физиотерапия и трудотерапия
В зависимости от потребностей вашего ребенка и того, насколько близко вы живете к больнице, дополнительные специалисты, включая хирургов, офтальмологов, стоматологов, кардиологов, эндокринологов, гематологов и другие, могут быть назначены на тот же день, за день до или на следующий день после родов. прием в клинику EB.
.