Лечение синдрома кошачьего крика — врачи, лечащие заболевание
Марина Юрьевна внимательный и профессиональный врач. Она предложила нам лечебное учреждение куда нужно ещё обратиться и выписала средства.
Светлана,
20 мая 2021
Хороший и честный специалист. Он знает как лечить человека. Я второй раз у него на приеме. Врач назначает мне лекарства.
Жасгуль,
18 мая 2021
В апреле обратилась к доктору в клинике ‘Евромедклиник 24’ в Жулебино. Опыт сугубо отрицательный. Из плюсов: сама клиника действительно хорошая и врач была адекватная (не кричала, не истерила, в принципе контактировала на нормальном уровне и вежливо).
Жалоба с которой я пришла: недавние боли в шейном отделе позвоночника, в течение месяца периодические головокружения (похожие на ДППГ), периодические ежедневные боли в нижних и верхних конечностях.
Осмотр был общий, спец.осмотра на предмет головокружения проведено не было. Есть сомнения в должном профессионализме врача и обладает ли она нужным объёмом умений и знаний. Обращаю внимание, у специалиста стоит в характеристике, что ‘Доброкачественное позиционное головокружение’ — тоже её профиль! В итоге у меня она его несмотря на идеальные симптомы не нашла, а нашла абстинентный синдром от отказа от курения спустя более месяца после полного отказа. Не уверена в правдивости описания её опыта и профессионализма. Спасибо, что не стало хуже под ведением этого специалиста, но лучше не стало однозначно.
Обращаю внимание, у специалиста стоит в характеристике, что ‘Доброкачественное позиционное головокружение’ — тоже её профиль! В итоге у меня она его несмотря на идеальные симптомы не нашла, а нашла абстинентный синдром от отказа от курения спустя более месяца после полного отказа. Не уверена в правдивости описания её опыта и профессионализма. Спасибо, что не стало хуже под ведением этого специалиста, но лучше не стало однозначно.
Постановка диагноза: неверная, по причине я уж не знаю чего. У меня симптомы и тип головокружения были прямо как по учебнику на ДППГ, которое, кстати, в итоге и оказалось, диагностированное не Олесей Валентиновной, а мной лично по интернету. Дома же сама сделала недавно манёвр Эпли и его однократное применение полностью сняло все мои симптомы головокружения. За один раз! Так что я не голословно на врача ‘гоню’.
Олеся Валентиновна же меня радостно собиралась лечить от последствий отказа от курения сосудистых, которые, по её мнению, вызывали головокружения. Возможная версия, если бы я бросила курить не за полтора месяца до визита к неврологу, а вот только что. Но Олеся Валентиновна была непреклонна, прописала мне месяц Мексидола, месяц Мильгаммы и немножко попить НПВС (который я сама же себе и выбрала, тк врач эту тему не знает также), чтобы снять боль в шее. Ну и ‘на всякий случай попробуйте сделать рентген шеи, может быть увидим что-то’. В общем, товарищи, это труба.
Возможная версия, если бы я бросила курить не за полтора месяца до визита к неврологу, а вот только что. Но Олеся Валентиновна была непреклонна, прописала мне месяц Мексидола, месяц Мильгаммы и немножко попить НПВС (который я сама же себе и выбрала, тк врач эту тему не знает также), чтобы снять боль в шее. Ну и ‘на всякий случай попробуйте сделать рентген шеи, может быть увидим что-то’. В общем, товарищи, это труба.
Врач в принципе была рассеяна, внимательно не слушала, реакций на рассказ никаких не выдавала, анамнез собирала по знакомым словам и явлениям, а не по картине клинической. Во время сбора анамнеза, когда врачу нужно быть максимально внимательным к рассказу пациента, в кабинет спокойно входила раза 2 или 3 какая-то сотрудница, что-то брала и уходила. Олеся Валентиновна на это апатично смотрела, будто всё в порядке, отвлекаясь от моего рассказа и, собственно, у меня вообще есть сомнения, что меня слышали.
Сбор анамнеза. По итогам моего изначального рассказа и осмотра у врача не возникло никаких предположений даже, на лице чистый лист и тишина в ответ. Я осторожно сообщила, что, да, кстати между прочим, полтора месяца назад я также бросила курить. В эту же минуту врач радостно сказала, что ‘ну это же очевидно! Вы бросили курить, поэтому сосуды перестраиваются, от этого и головокружения у Вас!’.
Я осторожно сообщила, что, да, кстати между прочим, полтора месяца назад я также бросила курить. В эту же минуту врач радостно сказала, что ‘ну это же очевидно! Вы бросили курить, поэтому сосуды перестраиваются, от этого и головокружения у Вас!’.
Назначение лечения. Врач после своего озарения начала с энтузиазмом выписывать мне инъекционное лечение, на что я сообщила, что уколы не приемлю. Колоть сама не буду, колоть мне дома некому, а ходить постоянно платно колоть в клинику я тоже пас. Уточнила у неё, есть ли аналоги в таблетках (и да, я знаю и знала тогда, что инъекции эффективнее) и врач мне согласилась выписать препараты в иной форме. Правда, чере3 минуты забыла уже об этом и снова бодро выписывала уколы. Прелестная дама.
В рамках беседы Олеся Валентиновна хотела прописать мне никотиновую кислоту вдобавок к тому, что уже было предписано ранее, но в итоговом назначении про неё — ни слова. Видимо, не прошла фэйсконтроль. Иных причин не вижу для того, чтобы за три минуты забыть о назначении.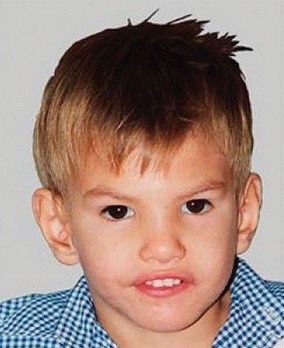
Часть назначенных микроэлементов для приема написаны были почему-то не на бланке, а на микро-листочке отрывном, что для меня тоже странно. На вопрос ‘в какой дозировке пить и как часто’ ответ был ‘ой, ну купите, там посмотрите всё’. Классно.
Яна,
12 мая 2021
Доктор внимательный, душевный, грамотный, квалифицированный и человечный врач. Она нам всё по полочкам разложила, объяснила и оказала необходимую помощь, которую мы ожидали месяц. Мы очень благодарны ей!
На модерации,
24 мая 2021
Хочу выразить огромную благодарность Орлову Григорию Анатольевичу.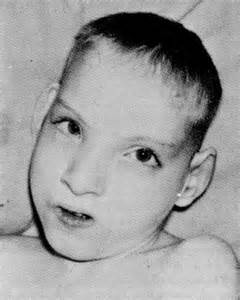
Переболев в тяжёлой форме ковидом, случилось обострение головного мозга, как это у многих и часто бывает.
Обратилась к доктору, пройдя МРТ.
Григорий Анатольевич очень внимательно выслушал , задал несколько встречных вопросов и назначил Лечение, которое длилось два месяца.
Я почувствовала себя значительно лучше.
Доктор очень внимательный, доброжелательный и профессионал своего дела.
Здоровья вам Григорий Анатольевич на долгие годы.
Побольше нам таких докторов!
С уважением Хромова Н. Г 24 мая 2021 г
Г. Москва
На модерации,
24 мая 2021
Синдром кошачьего крика
Содержание статьи
Причины
Спровоцировать изменения на генном уровне могут факторы, оказывающие негативное воздействие на репродуктивные клетки родителей. При этом стоит учитывать и отрицательное влияние уже на оплодотворенную яйцеклетку в период образования зиготы. Установлено, что к главным причинам относят:
При этом стоит учитывать и отрицательное влияние уже на оплодотворенную яйцеклетку в период образования зиготы. Установлено, что к главным причинам относят:
- употребление спиртного;
- наследственность;
- взаимодействие с токсичными веществами;
- курение;
- прием определенных лекарственных препаратов;
- наркомания.
Согласно проведенным исследованиям главная причина возникновения недуга – наследственный фактор. Если у одного из членов семьи имеется хромосомная болезнь, на этапе подготовки планирования беременности стоит пройти полное генетическое обследование.
Симптомы
Явный признак заболевания – плач малыша. У других малышей он переходит в хроническую форму. Также наблюдаются сопутствующие патологии:
- умственная отсталость;
- небольшой вес при рождении;
- уменьшение мышечного тонуса;
- пороки сердца;
- агрессия;
- задержка развития речи;
- гиперактивность;
- повышенное слюноотделение;
- регулярные запоры;
- нарушение функции глотания.

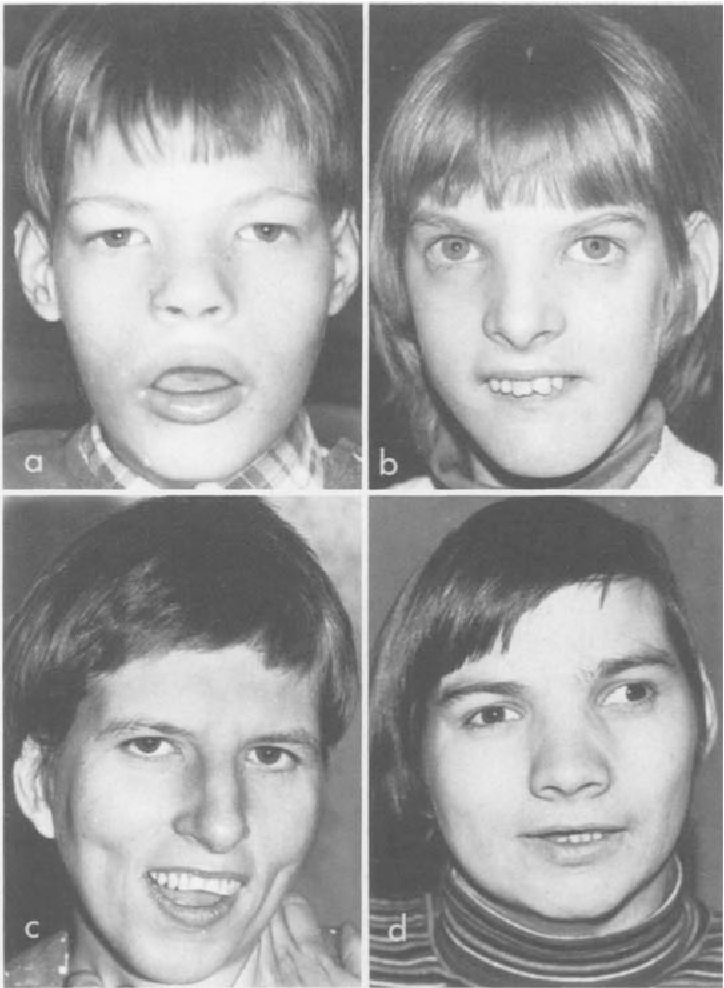
Кроме вышеуказанных проявлений, выделяют еще внешние признаки, такие как круглое лицо с широко расположенными глазами, плоская переносица, широкий нос, уши расположены низко, короткая шея.
Часто у ребенка, страдающего этим заболеванием, присутствуют и другие болезни, такие как расщепление губы и неба, косоглазие, атрофия зрительного нерва, непроходимость кишечника, гидронефроз, высокая гибкость суставов, плоскостопие и др.
Возрастное созревание и регулярные менструальные циклы у девочек проходят вовремя. У мальчиков наблюдается маленький размер тестикул.
При наблюдении одного из перечисленных проявлений необходима консультация генетика.
Диагностика
Доктор проводит первичный осмотр, направляет на кардиологическое обследование с использованием одной из методик идентификации хромосом. Для подтверждения диагноза нужно выполнить инвазивную пренатальную диагностику. В случае наличия изменений короткого плеча пятой хромосомы у крохи родители должны пройти цитогенетическое исследование.
Лечение
В основе терапии лежит устранение сопутствующих заболеваний, а также поддержание жизненно важных функций организма. Это связано с тем фактом, что в настоящее время данная патология неизлечима. Согласно статистическим показателям, только 10% малышей с таким недугом доживает до десяти лет. Чаще всего лечение предполагает выполнение хирургического вмешательства.
Для корректировки психомоторной деятельности назначается прием медикаментов и психотерапия. Массаж и лечебная физкультура эффективно влияют на улучшение состояния больного.
Профилактика
Чтобы не допустить развития патологии, доктора рекомендуют при планировании беременности пройти обследование, отказаться от вредных привычек.
Особенности лечения синдрома кошачьего крика. Симптомы, признаки и лечение синдрома Лежена (кошачьего крика) Синдром кошачьего крика вызывается мутацией
Синдром кошачьего крика (или синдром Лежена) — генетическое заболевание, которое встречается очень редко и связано с тем, что отсутствует часть 5 хромосом.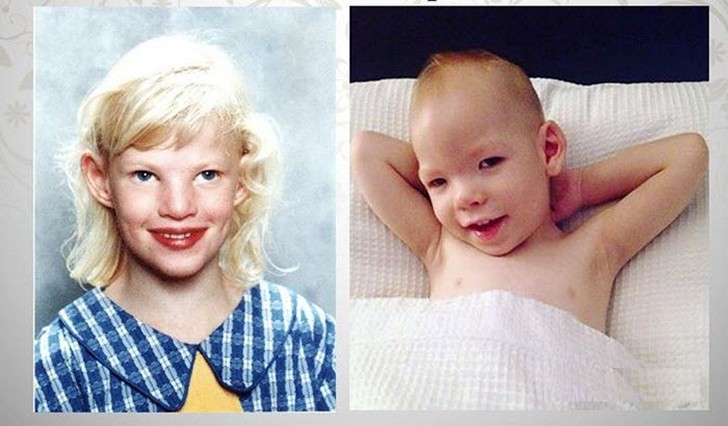 Дети, страдающие от этого заболевания, часто плачут, причем их плач похож на крик кошки. Отсюда и появилось название — синдром кошачьего крика.
Дети, страдающие от этого заболевания, часто плачут, причем их плач похож на крик кошки. Отсюда и появилось название — синдром кошачьего крика.
Это синдром возникает у одного ребенка на 50000 родившихся. Бывает в любой чаще поражает женский пол.
Впервые описал это заболевание французский генетик и педиатр Жером Лежен. Случилось это в 1963 году. Отсюда и второе название заболевания.
Симптомы заболевания
Синдром кошачьего крика возникает из-за определенных проблем с нервной системой и гортанью. По причине таких проблем появляется крик малыша, очень похожий на тот, что издают кошки. Примерно одна треть детей с этим синдромом теряют характерную черту (крик) уже к двум годам.
Симптомы, которые могут свидетельствовать о том, что у ребенка синдром кошачьего крика:
Трудности с питанием, особенно это касается сосании и глотания;
Маленький вес малыша и медленное физическое развитие;
Задержка развития речевых, когнитивных функций и функций движения;
Поведенческие проблемы: агрессия, гиперактивность и истерики;
Нетипичные черты лица, которые по прошествии времени могут исчезнуть;
Чрезмерное слюновыделение.
Кроме того, типичными признаками заболевания можно назвать: гипотонию, микроцефалию, задержку в развитии, круглую форму лица, опущенные уголки глаз, плоскую спинку носа, косоглазие, уши, расположенные слишком низко, короткие пальцы и прочее. Люди с синдромом Лежена чаще всего не имеют никаких проблем с репродуктивной системой.
Диагностика заболевания
Обычно диагноз ставят на основании характерного для данного симптома крика и прочих симптомов, перечисленных выше. Кроме того, семьям, где уже есть люди, страдающие от этой болезни, могут предложить генетическое тестирование и консультации на тему того, какими могут быть синдромы беременности.
К чему ведет синдром?
К сожалению, прогнозы для человека, страдающего от синдрома кошачьего крика, довольно неутешительны. Ведь продолжительность их жизни значительно меньше, чем у здоровых людей. Причем больные могут погибать не только от самого синдрома, но и от осложнений, которые его сопровождают (почечная и сердечная недостаточности, инфекционные заболевания).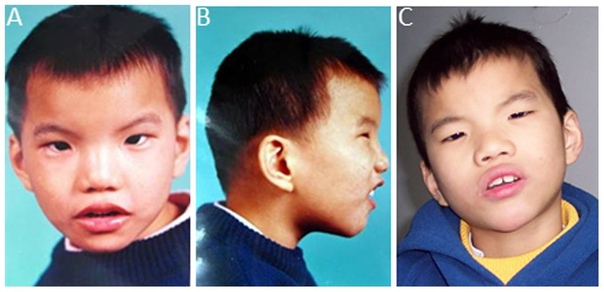
Клиническая картина и срок жизни больного может довольно сильно варьировать. Все зависит от того, насколько сильно поражены внутренние органы, особенно это касается сердца.
Больным делают массажи, гимнастику, прописывают лекарственные препараты, которые стимулируют
Если хотите избежать ситуации рождения ребенка, страдающего от такого заболевания, как синдром кошачьего крика, ваша обязательно должна пройти генетическое обследование и получить консультацию специалиста. Только так вы можете обезопасить себя от рождения неизлечимо больного ребенка. А это мечта каждой семейной пары, не так ли?
)
врожденный комплекс пороков развития, обусловленный нарушением структуры одной из хромосом группы В. Описан в 1963 г. Леженом с соавторами.
Частота синдрома среди новорожденных около 1: 3000, мальчики и девочки поражаются одинаково часто. Зависимость частоты рождения детей с Л.с. возраста родителей не установлена. обусловлен изменениями короткого плеча 5-й пары (рис. 1
1
), возникающими чаще вследствие участка хромосомы, реже — структурной перестройки хромосомы или перемещения сегмента хромосомы внутри хромосомного набора. Описаны и другие варианты сбалансированных транслокаций в клетках родителей, приводившие к рождению детей с синдромом Лежена. Некоторая вариабельность клинических проявлений синдрома, по-видимому, зависит от размеров недостающего участка хромосомы.
Дети с Л.с. обычно рождаются с низкой массой тела (до 2500 г
) даже при доношенной беременности. Наиболее постоянным признаком является специфический плач, напоминающий кошачье мяуканье. Этот обусловлен особенностью строения гортани, определяемым при ларингоскопии — маленьким вялым надгортанником, который может опускаться голосовой щелью. Голосовые складки не изменены. Рентгенологически отмечается уменьшение воздушного пространства над голосовыми складками.
В раннем детском возрасте характерны лунообразное , косой глаз с опущенными наружными углами, (складка у внутреннего угла глаза), (широко расставленные глаза), несколько уплощенный , низко расположенные ушные раковины (рис.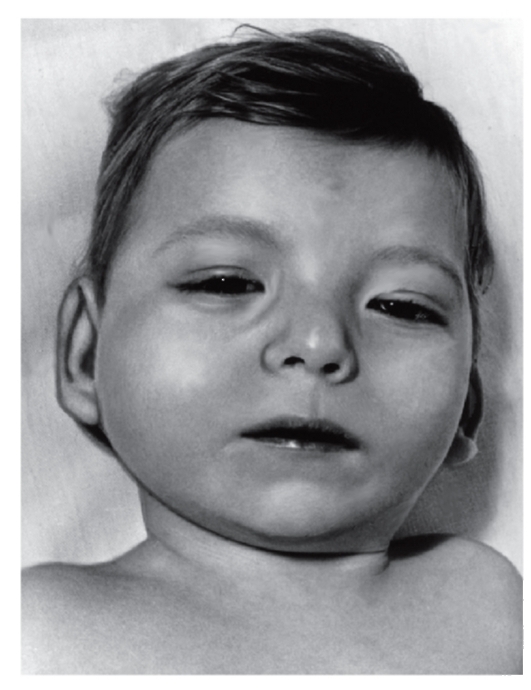
2
), впереди которых часто имеются небольшие (размером 1-3 мм
) круглые фиброзные узелки. Мозговой относительно малых размеров (), долихоцефальной формы (значительное преобладание продольных размеров над поперечными) или с выступающими лобными буграми. Отмечается маленькая нижняя челюсть и короткая с избыточной кожей, формирующей крыловидные складки. В некоторых случаях отмечается верхней или неба либо высокое готическое небо и расщепление язычка. Возможны преходящее или постоянное Косоглазие ,
астигматизм (см. Рефракция глаза).
В ряде случаев выявляют изменения глазного дна, в частности очаги депигментации сетчатки, а также атрофию зрительного нерва. Из аномалий развития внутренних органов наиболее часты сердца и сосудов, почек. У мальчиков часто бывает Гипоспадия .
Может быть четырехпалость или короткая, треугольной формы средняя фаланга V пальца. Общая мышечная , характерная для новорожденных с Л.с., обычно сохраняется в течение 1 года и дольше. У всех детей с Л.с. наблюдается умственная отсталость, большинство отстают в физическом развитии. Биохимические нарушения при Л.с. неспецифичны: длительное сохранение фетального гемоглобина, некоторое снижение содержания альбумина в сыворотке крови, умеренная и аминоацидурия.
У всех детей с Л.с. наблюдается умственная отсталость, большинство отстают в физическом развитии. Биохимические нарушения при Л.с. неспецифичны: длительное сохранение фетального гемоглобина, некоторое снижение содержания альбумина в сыворотке крови, умеренная и аминоацидурия.
Частота и выраженность отдельных признаков Л.с. имеют возрастную зависимость. Такие признаки, как плач, напоминающий кошачье мяуканье, мышечная гипотония, лунообразное лицо в большинстве случаев с возрастом полностью исчезают, а микроцефалия, косой разрез глаз становятся более выраженными; прогрессирует отставание в психомоторном развитии. Может быть стридор;
больные подвержены заболеваниям верхних дыхательных путей.
Синдром дифференцируют с другими врожденными пороками развития хромосомной и нехромосомной этиологии. подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом.
Лечение симптоматическое. Показаны средства, стимулирующие психомоторное развитие, лечебный и гимнастика.
Средняя продолжительность жизни больных снижена. Они погибают вследствие сердечной недостаточности (Сердечная недостаточность) или почечной недостаточности (Почечная недостаточность),
от интеркуррентных инфекционных болезней.
Профилактика заключается в своевременном проведении медико-генетического консультирования (Медико-генетическое консультирование) в семьях, где имелись больные с синдромом Лежена и основывается на определении кариотипа родителей, у которых был ребенок. Наличие изменений короткого плеча 5-й пары хромосом является абсолютным показанием для антенатального определения кариотипа плода при последующих беременностях путем амниоцентеза и исследования амниотических клеток. Сбалансированная у одного из родителей требует также исследования кариотипа у его кровных родственников с целью выявления лиц, имеющих транслокацию.
Библиогр.:
Козлова С.И. и др. Наследственные синдромы и медико-генетическое консультирование, с. 337, М., 1987; Маринчева Г. С. и Гаврилов В.И. Умственная отсталость при наследственных болезнях, с. 180, М., 1988; человека, под ред. Г.И. Лазюка, с. 314, М., 1979.
С. и Гаврилов В.И. Умственная отсталость при наследственных болезнях, с. 180, М., 1988; человека, под ред. Г.И. Лазюка, с. 314, М., 1979.
Рис. 1. Хромосомный набор больной с синдромом Лежена: групповая (от А до G) и индивидуальная идентификация хромосом (стрелкой указан дефект короткого плеча хромосомы 5-й пары, вторая хромосома не изменена).
II
Леже́на синдро́м (J. Lejeune, р. 1926 г., франц. педиатр и генетик)
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991-96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982-1984 гг
.
Синдром кошачьего крика или синдром Лежена относится к хромосомному виду заболевания, оно появляется тогда, если нет одной пятой хромосомы. Французский педиатр Ж. Лежен детально исследовал данное заболевание. При нем новорожденный имеет необычный , который похож на мяуканье кота.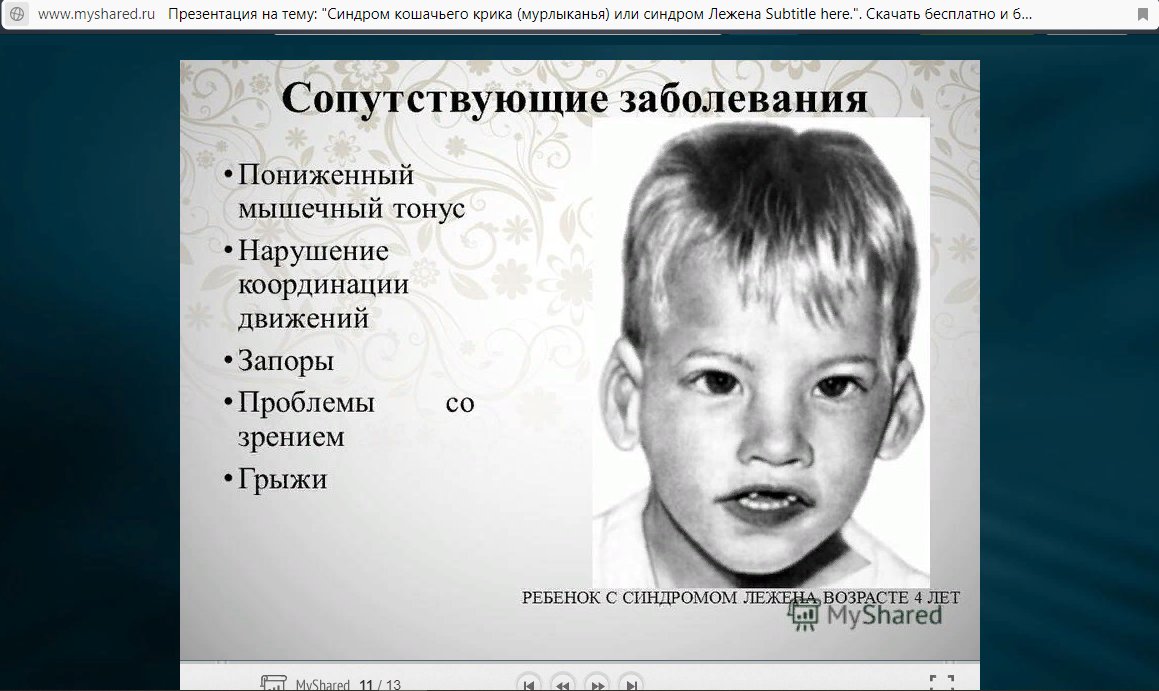 Синдром Лежена редкое заболевание, чаще всего возникает у девочек. Специального лечения нет, чаще всего используют хирургический метод, с помощью которого устраняются аномалии и разные нарушения.
Синдром Лежена редкое заболевание, чаще всего возникает у девочек. Специального лечения нет, чаще всего используют хирургический метод, с помощью которого устраняются аномалии и разные нарушения.
Причины синдрома кошачьего крика
Много факторов могут воздействовать на половой клеточный уровень, яйцеклетку, чаще всего заболевание спровоцировано:
1. Наследственными причинами, когда в семье кто-то имел такое заболевание.
2. Вредными привычками. Когда беременная женщина злоупотребляла алкогольными напитками и много курила.
3. Приемом , из-за них внутренние органы постепенно разрушаются, затем поражается генетический клеточный аппарат.
4. Воздействием радиации.
3. Употреблением сильнодействующих медикаментозных препаратов, которые женщина принимает в начале беременности.
Основные симптомы синдрома кошачьего крика
Дети с данным заболеванием рождаются своевременно, женщина до конца вынашивает ребенка, но он имеет небольшую массу тела, в среднем около 2500 грамм. Беременность протекает хорошо, патологических процессов при ней не наблюдается.
Беременность протекает хорошо, патологических процессов при ней не наблюдается.
Ребенок плачет так, как мяукает кошка, это сразу заметно. Такой звук объясняется тем, что малыш имеет своеобразную анатомическую гортанную особенность. В гортани просвет является узким, а хрящи размягченные. Дети при рождении имеют небольшой надгортанник. Часто синдром проходит уже в 2 года, опасно, когда заболевание остается на всю жизнь.
Синдром кошачьего крика и аутизм
Когда врач начинает исследовать геному семьи, он узнает, что в роду у ребенка был больной аутизмом. Если у кого-то из членов семьи были аномальные явления в хромосомном наборе, заболевание будет прогрессировать.
Дети, у которых синдром Лежена отличаются лунообразным лицом. При синдроме деформируются ушные раковины, укорачивается шея. Больной ребенок имеет заболевание – , у него проблемы с рефлексами, наблюдается гипотония в мышцах, нарушается сосание груди, глотание молока.
Иногда больной синдромом кошачьей крик имеет серьезные глазные заболевания – , проблемы со зрительным нервом, катаракту. У ребенка наблюдаются патологические процессы в опорно-двигательном аппарате – вывихнуто бедро, грыжа паха и пупка, синдактилия стоп. Опасно, когда синдром сопровождается заболеваниями почек, сердца, сосудов, желудка, кишечника. Дети, у которых синдром кошачий крик гиперактивны, страдают от истерии, агрессивны. Часто у них возникают проблемы с физическим развитием, ребенок умственно отсталый, имеет проблемы с речью.
У ребенка наблюдаются патологические процессы в опорно-двигательном аппарате – вывихнуто бедро, грыжа паха и пупка, синдактилия стоп. Опасно, когда синдром сопровождается заболеваниями почек, сердца, сосудов, желудка, кишечника. Дети, у которых синдром кошачий крик гиперактивны, страдают от истерии, агрессивны. Часто у них возникают проблемы с физическим развитием, ребенок умственно отсталый, имеет проблемы с речью.
Часто больные дети живут недолго, из-за того, что синдром Лежена сопровождается серьезными патологическими процессами в жизненно важных органах. Реже ребенок может дожить до 15 лет, и только 5% может жить с заболеванием около 50 лет.
Диагностика синдрома кошачий крик
Если в семье у кого-то наблюдается генетические заболевания, важно сдать специальный тест до того, как планируете беременность. Гинеколог во время беременности проводит дополнительное обследование, если УЗИ показывает аномалии. Диагноз можно подтвердить с помощью амниоцентеза, биопсии хориона, кордоцентеза.
После того как ребенок рождается и у него обнаружен синдром Лежена, его осматривает офтальмолог, кардиолог, ортопед, уролог, другие врачи.
Лечение синдрома Лежена
Современная медицина до сих пор не имеет специальных методов лечения хромосомного заболевания. Курс терапии может быть симптоматическим, он помогает поддержать в норме состояние системных органов и продолжить жизнь больного.
Чтобы наладилась психомоторная функция у ребенка, нужно обратиться к неврологу, пройти дополнительно медикаментозную терапию, дополнительно нужна консультация психолога. Дополнительно ребенка нужно показать логопеду, физиотерапевту, дефектологу.
Если ребенок родился с кошачьим синдромом, имеет врожденные , необходима срочная операция. Часто синдром сопровождается аномалиями в мочевыделительной системе, поэтому сразу нужно обращаться к урологу.
Из-за того, что у ребенка общий гипотонус, ему нужен постоянный массаж, лечебная гимнастика. Врачи выписывают лекарственные препараты, с помощью которых происходит стимуляция психомоторного развития.
Прогноз при синдроме кошачьего крика
Сколько будет жить ребенок, зависит от того, насколько повреждены хромосомы. Очень редко у человека все нормально и он доживает до 60 лет. Если заниматься ребенком, можно его научить чтению, писанию, адаптироваться к окружающей среде.
Продолжительность жизни с данным заболеванием зависит от того, какие врожденные пороки имеет ребенок, оказана ли своевременно медицинская, психологическая помощь. Часто, если заниматься с ребенком, он будет иметь в словарном запасе несколько предложений.
Профилактика синдрома Лежена
Необходимо тщательно готовиться к беременности, сдать все необходимые анализы, чтобы исключить все генетические причины, также беречься во время беременности, отказаться от всех вредных привычек, защищать себя и ребенка от всех неблагоприятных факторов. Если в семье рождается ребенок с синдромом кошачий крик, необходимо дополнительно сдать цитогенетический анализ, чтобы исключить все осложнения.
Итак, синдром кошачьего крика – это редкое, но опасное заболевание. Лучше предотвратить его, потому что вылечить невозможно. Будущая мама должна тщательно относиться к своему здоровью, забыть за алкоголь, курение. Если ребенок родился с аномалиями, тщательно следить за его здоровьем, обратить, полностью обследовать его, потому что часто синдром сопровождается разными патологиями в органах.
Лучше предотвратить его, потому что вылечить невозможно. Будущая мама должна тщательно относиться к своему здоровью, забыть за алкоголь, курение. Если ребенок родился с аномалиями, тщательно следить за его здоровьем, обратить, полностью обследовать его, потому что часто синдром сопровождается разными патологиями в органах.
Синдром кошачьего крика относится к редким генетическим заболеваниям, Хромосомным мутациям, аномалиям. При этих нарушениях целые хромосомы или большие их сегменты отсутствуют, дублируются или иным образом изменены.
Профессиональная справочная статья основана на британских и европейских медицинских научных данных.
Синдром Лежена /кошачьего крика, называемый также Cri-du-chat (Кри-дю-чат, CdCS), CHB, 5p- (5 Р минус), часовой, врожденное нарушение частичной аутосомной делеции (удаления) короткого плеча хромосомы 5.
Назван за его характерный пронзительный плач похожий на кошачий крик. Характеризуется ростовой недостаточностью, врожденными патологиями, общей нетрудоспособностью, умственной отсталостью на протяжении всей жизни.
Встречается во всех этнических группах. Был описан французским генетиком, педиатром Жером-Жан-Луи-Мари Леженом и его коллегами в 1963 году.
Кошачье мяуканье проходит по мере взросления. Клиническая тяжесть связанна с размером удаления.
Диагностируется у детей грудного и раннего возраста на основе клинических симптомов, подтверждается при помощи генетического анализа. Может быть обнаружен до рождения через пренатальное тестирование на образцах ткани плаценты или плода, собранных путем отбора проб хорионического ворсинок или амниоцентеза.
Причины
Синдром кошачьего крика/Лежена вызван частичной делецией (моносомой) различной длины короткой руки (p) хромосомы 5. Хромосомы, присутствующие в ядре клеток, несут генетическую информацию для каждого человека.
Пары пронумерованы от 1 до 22, еще 23-й пары половых, которые включают одну Х и одну Y-хромосому у мужчин и две Х- у женщин. Каждая имеет короткую руку, обозначенную «р», а длинную руку обозначают «q».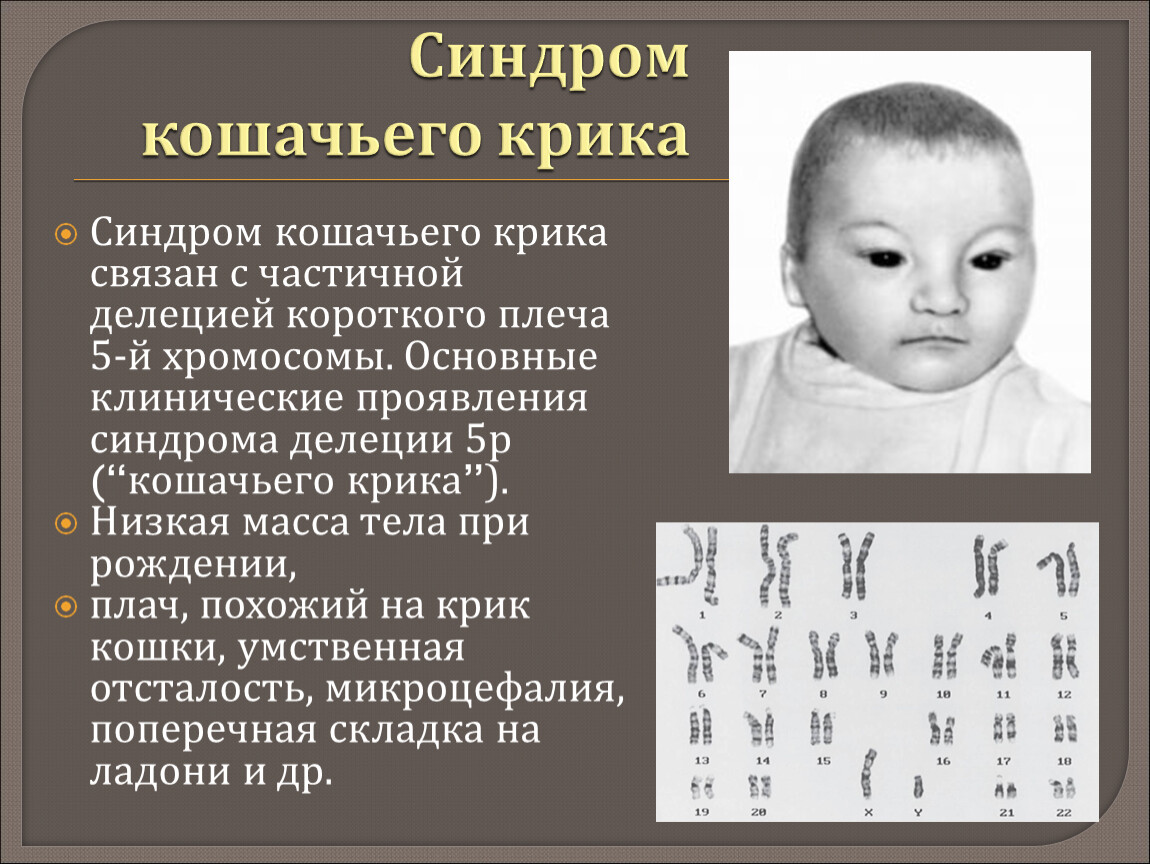
Они подразделяются на многие группы, которые нумеруются. Например, «5p15.3» относится к полосе 15. Пронумерованные полосы определяют местоположение тысяч генов. Диапазон и тяжесть связанных симптомов варьируется, зависит от конкретных затронутых участков.
Исследователи выявили несколько генов, которые, играют роль в Cri du Chat. Ген теломеразы обратной транскриптазы, расположеный на коротком плече хромосомы 5 полосы 13,33 (5,13,33) и ген семафорина F при 5p15.2 способствуют широкому разнообразию признаков.
Удаление гена d-catenin в 5p15.2, дает более тяжелую умственную недееспособность, поскольку этот белок экспрессируется при раннем формировании нейронов.
Большинство мутаций возникают спонтанно (de novo) по неизвестным причинам в самом начале эмбрионального развития.
Родители ребенка с делецией обычно имеют нормальные гены и относительно низкий риск появления другого малыша с патологией.
Примерно в 10-15% возникает из-за сбалансированной транслокации с участием 5р и другой хромосомы.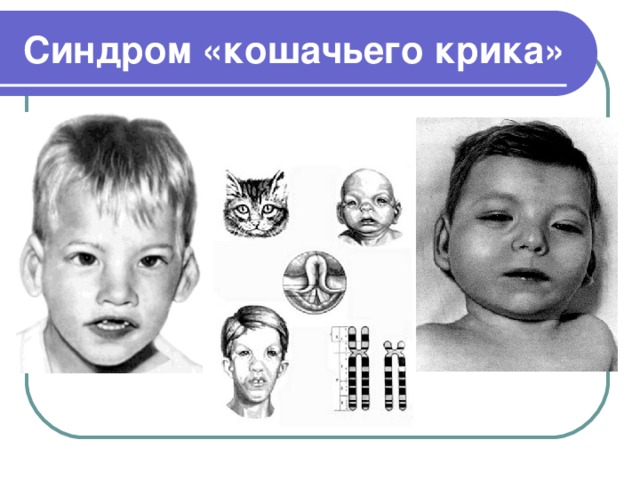 Транслокации происходят, когда области прерываются, перегруппированы, что приводит к смещению генетического материала, измененному набору. Такие транслокации появляются спонтанно по неизвестным причинам или передаются от носителя транслокации.
Транслокации происходят, когда области прерываются, перегруппированы, что приводит к смещению генетического материала, измененному набору. Такие транслокации появляются спонтанно по неизвестным причинам или передаются от носителя транслокации.
Транслокация состоит из измененного, но сбалансированного набора и безвредна для носителя. Однако, хромосомная перегруппировка повышает риск появления патологии у потомков. Генетический анализ определяет, имеет ли родитель сбалансированную транслокацию.
Интересные факты
В 80 процентах делеция, происходит от спермы отца, а не от материнской яйцеклетки.
Распространенность, эпидемиология
Сообщается, что заболеваемость составляет от 1 до 15 000 и 1 из 50 000 живорождений.
- Существует небольшое преобладание женщин с соотношением около 4: 3.
- Некоторые случаи не диагностируются, что затрудняет определение истинной частоты этого расстройства у населения в целом.
80% случаев представляют собой новые мутации, но приблизительно у 15% есть родитель со сбалансированной перегруппировкой. Остальные происходят из-за редких цитогенетических аберраций – например, мозаицизма.
Остальные происходят из-за редких цитогенетических аберраций – например, мозаицизма.
Симптомы и признаки
Из-за изменения размера удаления могут быть значительные колебания состояния.
Обратите внимание на гипертонус, маленькое, узкое лицо, челюсть с выпуклым днищем, а также выражение лица, вторичное по отношению к лицевой слабости
Младенцы малы при рождении, у них возникают проблемы с дыханием. Часто гортань развивается неправильно, что вызывает сигнальный звук.
Люди, получившие поражение кошачьего крика, имеют общие симптомы и очень отличительные черты.
У них может быть небольшая голова (микроцефалия), необычно круглое лицо, маленький подбородок, широко расставленные глаза, складки кожи на глазах, небольшая переносица.
Со временем, лицо может потерять свою полноту, стать ненормально длинным и узким
Наиболее распространенные признаки в младенчестве
- Характерный крик, как мяуканье кошки, дал заболеванию свое название. Генетические исследования подтвердили, что это результат делеции при 5p15.
- Продолжительность плача противоречива, но со временем исчезает. Около трети потеряли ее к второму дню рождения. Несмотря на то, что характерный звук очень распространен, нельзя сказать, что он патогномоничен, так как наблюдается при некоторых других неврологических расстройствах. В редких случаях присутствует постоянный стридор.

- Низкий вес при рождении (
- Проблемы с кормлением (плохой сосательный рефлекс, дисфагия, мышечная гипотония, ).
Синдром «кошачьего крика»
– хромосомное нарушение, обусловленное делецией (отсутствием) фрагмента короткого плеча 5-ой хромосомы. Плач новорожденных с синдромом «кошачьего крика» по звуку напоминает кошачье мяуканье, что и послужило названию патологии. Кроме этого, у детей имеет место микроцефалия, лунообразное лицо, косоглазие, аномалии прикуса, различные врожденные пороки, грубое интеллектуальное недоразвитие и т. д. Синдром «кошачьего крика» диагностируется на основании совокупности характерных признаков и цитогенетического исследования.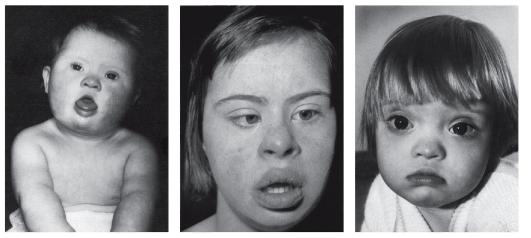 Специфического лечения синдрома «кошачьего крика» не существует; дети могут нуждаться в хирургической коррекции тяжелых врожденных аномалий.
Специфического лечения синдрома «кошачьего крика» не существует; дети могут нуждаться в хирургической коррекции тяжелых врожденных аномалий.
Синдром «кошачьего крика» (синдром Лежена) – частичная моносомия, связанная с нарушением структуры короткого плеча 5-ой хромосомы (потерей от 1/3 до 1/2 его длины, реже — полной утратой короткого плеча). Синдром «кошачьего крика» относится к числу редких хромосомных заболеваний с популяционной частотой 1:45-50 тыс. Среди новорожденных с синдромом «кошачьего крика» отмечается преобладание девочек над мальчиками в соотношении 4:3. Заболевание было описано в 1963 г. французским генетиком и педиатром Ж. Леженом и по автору получило название «синдром Лежена». Однако в литературе за данной патологией закрепилось образное название, связанное со специфическим признаком – плачем новорожденных, напоминающим кошачий крик.
Причины синдрома «кошачьего крика»
Развитие синдрома «кошачьего крика» связано с потерей фрагмента 5-ой хромосомы, а, следовательно, генетической информации, хранящейся на этом участке.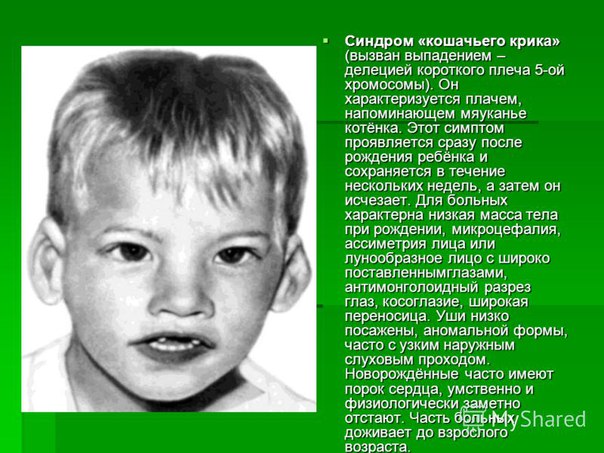 В 85-90% случаев делеция короткого плеча образуется в результате случайной мутации, в 10-15% наследуется от родителей, являющихся носителями сбалансированной транслокации.
В 85-90% случаев делеция короткого плеча образуется в результате случайной мутации, в 10-15% наследуется от родителей, являющихся носителями сбалансированной транслокации.
Наиболее частыми цитогенетическими вариантами хромосомной аберрации служат утрата одной трети или половины длины короткого плеча 5-ой хромосомы. Потеря меньшего участка или всего плеча встречается исключительно редко. При этом для степени выраженности клинической картины синдрома «кошачьего крика» важен не размер утерянного фрагмента, а отсутствие конкретного участка хромосомы. Так, при потере небольшого участка хромосомы в области 5p15.2 развиваются все клинические признаки синдрома, кроме кошачьего крика; критическим для возникновения характерного крика является выпадение участка хромосомы в области 5p15.3.
Наряду с простой делецией, могут встречаться другие цитогенетические вариации синдрома «кошачьего крика»: мозаицизм, кольцевая 5-я хромосома с делецией участка короткого плеча, реципрокная транслокация короткого плеча 5-ой хромосомы на другую хромосому.
Непосредственной причиной мутации могут выступать различные повреждающие факторы, воздействующие на половые клетки родителей либо на зиготу (алкоголь, курение, наркотические вещества, ионизирующая радиация, лекарственные препараты, химикаты и пр.). Вероятность появления ребенка с синдромом «кошачьего крика» выше в семьях, где уже рождались дети с подобным заболеванием.
Симптомы синдрома «кошачьего крика»
Новорожденные с синдромом «кошачьего крика», как правило, рождаются доношенными, но с небольшой пренатальной гипотрофией (средняя масса при рождении около 2500 г). Беременность у матери может протекать абсолютно нормально или сопровождаться угрозой самопроизвольного прерывания не чаще, чем в популяции. Наиболее патогномоничным ранним признаком синдрома является плач ребенка, который напоминает мяуканье кошки. Высокое и пронзительное звучание детского крика обусловлено анатомическими особенностями строения гортани при данном синдроме – узостью ее просвета, небольшим надгортанником, необычной складчатостью слизистой оболочки, мягкой консистенцией хрящей.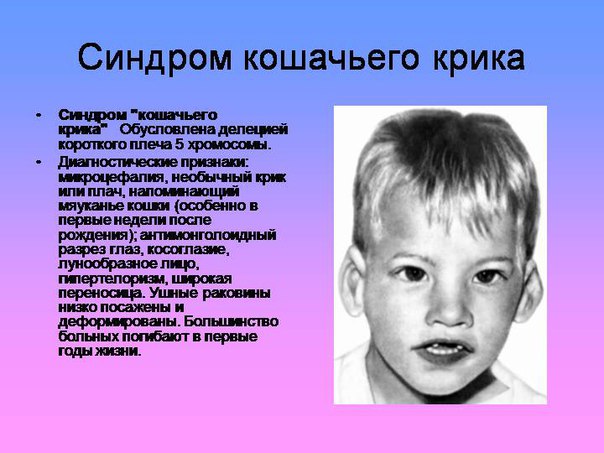 Некоторые авторы считают, что специфический крик имеет центральное происхождение и не связан с недоразвитием гортани. Примерно у трети детей «кошачий крик» исчезает к 2-м годам, у остальных остается на всю жизнь.
Некоторые авторы считают, что специфический крик имеет центральное происхождение и не связан с недоразвитием гортани. Примерно у трети детей «кошачий крик» исчезает к 2-м годам, у остальных остается на всю жизнь.
Фенотип детей с синдромом «кошачьего крика» отличается преобладанием лицевой части черепа над мозговой, лунообразным лицом, гипертелоризмом, антимонголоидным разрезом глаз, эпикантом, деформацией ушных раковин, плоской спинкой носа, короткой шеей с крыловидными складками. При обследовании у детей выявляется микроцефалия, мышечная гипотония, снижение рефлексов, нарушение сосания и глотания. В неонатальном периоде может развиваться инспираторный стридор и цианоз.
Другие клинические проявления синдрома «кошачьего крика» могут значительно варьироваться по своему сочетанию у отдельных больных. Со стороны зрительной системы нередко обнаруживается врожденная катаракта, близорукость, косоглазие, атрофия зрительного нерва. Изменения со стороны костно-мышечной системы проявляются синдактилией стоп, врожденным вывихом бедра, косолапостью, плоскостопием, клинодактилией V пальца кисти, сколиозом, диастазом прямых мышц живота, паховыми и пупочными грыжами. Частыми спутниками синдрома «кошачьего крика» являются нарушения прикуса, «готическое» нёбо, микрогения, расщелины нёба и верхней губы, расщепление язычка.
Частыми спутниками синдрома «кошачьего крика» являются нарушения прикуса, «готическое» нёбо, микрогения, расщелины нёба и верхней губы, расщепление язычка.
У многих пациентов наблюдаются врожденные пороки сердца (ДМЖП, ДМПП, открытый артериальный поток, тетрада Фалло), пороки развития почек (гидронефроз, подковообразная почка), крипторхизм, гипоспадия. Реже отмечается мегаколон, запоры, кишечная непроходимость. Дерматоглифическими признаками синдрома «кошачьего крика» могут служить одна ладонная складка, поперечные складки сгибания и др.
Поведение детей характеризуется гиперактивностью, однообразными движениями, склонностью к агрессии и истерикам. Детям с синдромом «кошачьего крика» свойственна глубокая умственная отсталость в степени имбецильности и идиотии, грубое системное недоразвитие речи, выраженное отставание в моторном и физическом развитии.
Половая и репродуктивная функции у лиц с синдромом «кошачьего крика» обычно не страдают. Иногда у женщин выявляется двурогая матка, у мужчин – уменьшение размеров тестикул, однако сперматогенез существенно не нарушен, как, например, при синдроме Клайнфельтера.
Продолжительность жизни больных с синдромом «кошачьего крика» значительно укорочена; большая часть детей погибает в первый год жизни из-за сопутствующих пороков и их осложнений (чаще от сердечной и почечной недостаточности). Лишь около 10% доживают до подросткового возраста, хотя имеются отдельные сообщения о больных, достигших 50 лет.
Диагностика синдрома «кошачьего крика»
Если в семье уже имелись случаи хромосомных заболеваний, еще на этапе планирования беременности будущим родителям рекомендуется посетить генетика и пройти генетическое тестирование. В процессе беременности наличие у плода синдрома «кошачьего крика» может быть заподозрено на основании результатов ультразвукового пренатального скрининга. В этом случае для окончательного подтверждения хромосомной аномалии рекомендуется проведение инвазивной пренатальной диагностики (амниоцентеза, биопсии ворсин хориона или кордоцентеза) и непосредственного анализа генетического материала плода.
После рождения предварительный диагноз синдрома «кошачьего крика» устанавливается неонатологом на основании типичных диагностических признаков (характерного плача, фенотипических черт, множественных стигм дизэмбриогенеза). Для подтверждения хромосомной патологии проводится цитогенетическое исследование.
Для подтверждения хромосомной патологии проводится цитогенетическое исследование.
Учитывая наличие у детей с синдромом «кошачьего крика» множественных аномалий развития, необходимо, чтобы в первые дни жизни новорожденные были осмотрены детским кардиологом, детским офтальмологом, детским урологом, детским ортопедом и другими специалистами.
Лечение синдрома «кошачьего крика»
Специфического лечения данного хромосомного заболевания в настоящее время не существует. Для стимуляции психомоторного развития под наблюдением детского невролога проводятся курсы медикаментозной терапии, массажа, физиотерапии, ЛФК. Дети с синдромом «кошачьего крика» нуждаются в помощи психологов, дефектологов, логопедов.
Врожденные пороки сердца при синдроме «кошачьего крика» часто требуют хирургической коррекции, поэтому детям необходима консультация кардиохирурга, проведение ЭхоКГ и других необходимых исследований. Дети с патологией мочевыделительной системы должны находиться под наблюдением детского нефролога и периодически проходить комплекс необходимых обследований (УЗИ почек, общий анализ мочи, биохимическое исследование крови и мочи и др. ).
).
Прогноз и профилактика синдрома «кошачьего крика»
На продолжительность и качество жизни больных влияет тяжесть самого синдрома и сопутствующих врожденных пороков, уровень оказания медицинской и психолого-педагогической помощи. В целом долговременный прогноз неблагоприятный. При специальном обучении дети имеют словарный запас, достаточный для бытового общения, однако по уровню психомоторного развития обычно не поднимаются выше дошкольников.
Профилактика синдрома «кошачьего крика» заключается в тщательной подготовке к беременности и исключении возможных неблагоприятных воздействий на организм родителей еще задолго до зачатия. При рождении в семье ребенка с синдромом «кошачьего крика», родители в обязательном порядке должны пройти цитогенетическое обследование для исключения носительства реципрокной сбалансированной транслокации.
Хромосомные нарушения
Хромосомные нарушения — это клинические синдромокомплексы, в основе которых лежат нарушения числа или структуры хромосом, то есть избыток или нехватка генетического материала, локализованного в той или иной хромосоме.
В норме у человека число хромосом равно 46, из которых 23 ребенок получает от матери и 23 аналогичные хромосомы от отца. В этом наборе гентического материала есть 2 особые хромосомы, которые были названы «половыми». Они определяют пол ребенка и ряд других важных признаков.
Таким образом, изменения числа хромосом (больше или меньше 46), а также изменение структуры хромосом (например, выпадение или удвоение даже небольшого кусочка хромосомы) получили название «хромосомные мутации».
Наиболее часто из них встречаются изменения модального числа хромосом — это отсутствие в хромосомном наборе какой-либо хромосомы (моносомия) или появление добавочной хромосомы (трисомия, тетрасомия и т.д.).
Число возможных изменений структуры хромосомы неисчислимое множество. К примеру, транслокации (обмен сегментами между разными хромосомами), делеции (выпадение участка хромосомы), дупликации (удвоение части хромосомы), инверсии (переворот сегмента хромосомы на 180 градусов) и т.д.
Хромосомные мутации, возникшие в половых клетках (сперматозоидах или яйцеклетках) или на первых этапах деления клеток зародыша, как правило, передаются большинству клеток развивающегося организма, вызывая множественные аномалии развития, а многие хромосомные изменения плода могут стать причиной спонтанных абортов и выкидышей, что важно учитывать в семьях, воспитывающих детей с задержками развития.
К факторам риска, способствующим их возникновению, относят ионизирующую радиацию, инфекции и интоксикации матери, эндокринные нарушения, психические травмы, воздействие ряда лекарственных препаратов и некоторых физиотерапевтических методов лечения.
Наиболее точно установлено, что причиной появления ребенка с хромосомными мутациями является не молодой возраст матерей (свыше 40 лет).
В последнее время очень большое значение придается факту скрытого носительства хромосомных нарушений у родителей родившегося ребенка (сбалансированные транслокации, мозаицизм). Изучение данного вопроса позволяет предотвратить риск повторного рождения ребенка с аналогичной формой заболевания.
Различают хромосомные синдромы, обусловленные изменением половых хромосом, и синдромы, вызванные аномалиями аутосом (любой из 44 неполовых хромосом).
Основными клиническими проявлениями аутосомных аномалий являются признаки психического и физического недоразвития, дисплазии (неправильное развитие), врожденные пороки развития (аномалии) и умственная отсталость различной степени тяжести. К врожденным порокам можно отнести: аномалии развития сердца, удвоение почки, расщелина неба, особенности строения кистей и стоп и многие другие. При заболеваниях, обусловленных нарушениями в системе половых хромосом, как правило, более характерны недоразвитие половых желез и аномалии развития вторичных половых признаков, также с симптомами задержки психо-речевого развития.
Различные хромосомные синдромы встречаются с разной частотой. По сводным данным многих исследований, распространенность наиболее частых из них среди новорожденных следующая:
трисомия по 21 хромосоме (синдром Дауна) 1:500
XXX (трисомия-Х) 1:1000 (девочек)
ХYY (синдром дубль-Y) 1:1000 (мальчиков)
ХХY (синдром Клайнфелтера) 1:1400 (мальчиков)
Х0 (синдром Шерешевского-Тернера) 1:3300 (девочек)
46,5р del (синдром «кошачьего крика») 1:4000
трисомия по 18 хромосоме (синдром Эдвардса) 1: 6800
трисомия по 13 хромосоме (синдром Патау) 1:7600
Не смотря на казалось бы не частую встречемость каждого отдельного синдрома, в целом хромосомные болезни у новорожденных наблюдаются не редко — с частотой около 1 : 100. Ежегодно в России рождается свыше 30 тыс. детей с хромосомной патологией. Спонтанные выкидыши являются результатом хромосомной патологии в более чем 50%.
Рассмотрим основные клинические проявления отдельных хромосомных синдромов, сопровождающихся умственной отсталостью и задержками психо-речевого развития.
Синдром Дауна — врожденное заболевание, характеризующееся умственной отсталостью и рядом признаков эндокринной недостаточности.
Синдром впервые описан английским врачом Дауном в 1866 г. Встречается с частотой 1 на 500 новорожденных. Частота встречаемсоти у мальчиков и девочек одинакова. В основе заболевания лежит аномалия хромосомного набора (47 вместо 46). Лишняя хромосома обнаруживается в 21 паре, в связи с чем этот синдром иногда называют «трисомией по 21-й хромосоме» (47, 21+). Выявлена связь частоты рождения больных с увеличением возраста матери. Приблизительно в 3—4% случаев отмечаются транслокационные формы синдрома Дауна, при которых общее число хромосом в кариотипе нормальное — 46, а дополнительная 21-я хромосома транслоцирована (присоединена) на другую аутосому. Это является результатом того, что один из фенотипически здоровых родителей является скрытым носителем сбалансированной транслокации. Именно за счет этих форм повышается риск повторного рождения больного ребенка у молодых матерей. Еще 3-4% случаев синдрома Дауна составляют мозаичные варианты, при которых в организме одновременно обнаруживают и трисомные, и нормальные клетки. Порой, при небольшом проценте трисомных клеток ребенок с ЗПРР внешне может выглядеть абсолютно нормальным.
Установлено, что для синдрома Дауна характерно уменьшение размеров и веса головного мозга, а также аномалии развития мозга и мозговых сосудов. Отмечаются также структурные изменения в железах внутренней секреции, печени и сердце. Клиническая картина синдрома Дауна характеризуется проявлениями симптомов умственной отсталости. Характерен также и внешний вид таких больных: косо расположенные глазные щели, широкая уплощенная переносица, дополнительная кожная складка у внутреннего угла глаз, высокое стояние твердого неба (признаки эмбриональной задержки в развитии лицевого скелета), полуоткрытый рот, увеличенный высунутый язык с выраженными сосочками и глубокими бороздами (признаки дисфункции щитовидной железы), выпадение волос (дисфункция надпочечников), низкий рост, короткая шея, укороченные кисти и стопы, искривление мизинца, на ладонях имеется поперечная складка, на стопах увеличен промежуток между 1 и 2 пальцами, выражены внешние проявления гипогенитализма.
Такие дети с рождения отстают в росте, начинают поздно держать голову, сидеть и ходить. Речь, как правило, невнятная, словарный запас беден, произношение с дефектами в связи с недоразвитием высших мозговых функций, с одной стороны, и анатомическими аномалиями ротовой полости — с другой.
В клинической картине заболевания доминируют симптомы неврологической патологии, диффузная мышечная гипотония (снижение мышечного тонуса), благодаря чему больные гибки и иногда могут складываться как «перочинный ножик», расстройства координации движений, косоглазие, выраженные вегетососудистые нарушения.
Особенностью психического дефекта является относительная сохранность эмоциональной сферы по сравнению с тяжестью интеллектуального недоразвития. Так, больные ласковы, добродушны, послушны. Характерной особенностью таких детей является повышенная внушаемость, что является положительным фактором при проведении коррекционной работы и отрицательным при их развитии.
Уровень социального развития больных с синдромом Дауна зависит от степени и формы заболевания. Так, дети с более легкими формами умственной отсталости, хотя и медленно, но развиваютя, приобретая определенные навыки, знания, осваивая программу нескольких классов вспомогательной школы. Однако, как правило, большинство из них не достигают удовлетворительного уровня социальной адаптации и нуждаются в постоянной опеке. Им может быть оформлена инвалидность детства с момента точной диагностики заболевания. Особенностью возрастной динамики синдрома Дауна является позднее половое созревание и раннее появление признаков инволюции (25—30 лет). Мужчины с синдромом Дауна бесплодны, женщины могут давать потомство, половина которого также страдает синдромом Дауна.
Синдром Шерешевского—Тернера — симптомокомплекс проявлений врожденного, наследственно обусловленного недоразвития половых желез и передней доли гипофиза в сочетании с аномалиями соматического развития.
Впервые заболевание описано отечественным эндокринологом Н.А. Шерешевским (1925), а более подробно — американским эндокринологом Н. Тернером (N.H. Terner) л 1938 г. В основе заболевания лежит отсутствие одной хромосомы (половой Х-хромосомы) (45 вместо 46).
Клиническая картина синдрома характеризуется разной степенью умственной отсталости и ЗПРР, низким конечным ростом (135—145 см), замедлением полового развития, недоразвитием половых желез, аменореей, бесплодием и отсутствием грудных желез. Диспластические расстройства проявляются в виде короткой шеи и особых кожных складок, идущих от затылка к надплечью, укорочением 4 пальцев на руках и искривлением мизинцев, выраженной деформацией ушных раковин, наличием множественных пигментных родинок. Преимущественно данным синдромом страдают лица женского пола.
Синдром Клайнфелтера — заболевание, обусловленное нарушением числа половых хромосом (добавочные Х-хромосомы) (от 47 до 49), характеризующееся умственной отсталостью, нарушением смерматогенеза, недоразвитием яичек и вторичных половых признаков, а также нарушением пропорций тела. Впервые синдром описан американским эндокринологом Клайнфелтером (H.F. Klinfelter) в 1942 г. Его частота, по сводным данным, составляет до 2% среди умственно отсталых и до 0,5% (кадждый двухсотый мужчина) в среднем в мужской популяции.
Клинические проявления синдрома Клайнфельтера варьируют от внешне нормального и интеллектуального развития до выраженного евнухоидизма и умеренной умственной отсталости. Однако в ряде случаев уже в раннем возрасте у больных отмечаются характерные своеобразные симптомы физического развития: низкий и узкий лоб, густые и жесткие волосы, высокое стояние таза, короткая, плоская и узкая грудная клетка, недоразвитие половых органов. Более отчетливо вышеперечисленные симптомы начинают обнаруживаться в подростковом, пубертатном возрасте. Характерен внешний вид взрослого больного с синдромом Клайнфельтера: высокий рост, астеническое сложение, узкие плечи, широкий таз, удлиненные конечности, слаборазвитая мускулатура, скудная растительность на лице и в подмышечных впадинах, ожирение и оволосение по женскому типу, сутулость, выраженные евнухоидные пропорции и гинекомастия (набухание грудных желез). Постоянными признаками синдрома Клайнфельтера являются недоразвитие половых органов и бесплодие.
Степень интеллектуального недоразвития у больных выражена тем глубже, чем больше дополнительных половых хромосом обнаруживается в кариотипе (46 или 49). Так, умеренная умственная отсталость зачастую приближается к психическому инфантилизму, что клинически проявляется недостаточностью внимания, восприятия, памяти, абстрактного мышления, чрезмерной внушаемостью, подражательностью, подчиняемостью, несамостоятельностью, чрезмерной привязанностью к близким, нередко с элементом назойливости. Глубокая незрелость эмоционально-волевой сферы проявляется в виде повышенного настроения, с эйфорическим оттенком, склонностью к эксплозивным аффективным вспышкам, неспособностью к длительному волевому усилию и напряженной деятельности. У больных, как правило, отсутствуют чувство долга и ответственности. При легких формах заболевания больные осознают свою неполноценность, что приводит к внутреннему конфликту и возникновению у них невротических реакций. Данным синдромом страдают лица мужского пола.
Синдром ломкой Х-хромосомы (Fragile X syndrome, FraХ). Начиная с 1980 года большое значение придают синдрому ломкой Х-хромосомы (Хq27.3) – именно с ним связывают развитие более чем 50 наследственных расстройств, включая ранний детский аутизм и 30% случаев умственной отсталости у мальчиков. Хрупкий участок Х-хромосомы впервые обнаружил Labs (1969).
Полная мутация в Х-хромосоме возникает только у женщин, и происходит это в процессе гаметогенеза, поэтому почти всегда страдают мальчики, получившие единственную Х-хромосому от матери. У девочек, получивших вторую Х-хромосому от отца, также могут быть нарушения развития, но они менее выражены, а тяжелые патологии встречаются много реже, чем у мальчиков. В отдельных случаях девочки могут получить обе ломкие хромосомы от матери, в этом случае частота и тяжесть патологии будет одинаковой с мальчиками.
Клиническую триаду синдрома ломкой Х-хромосомы образуют:
1) умеренная до степени тяжелой умственная отсталость. Лишь 30% лиц мужского пола имеют интеллект, стремящийся к нижней границе нормы, а среди женщин – носительниц такой хромосомной патологии примерно у 30% обнаруживаются признаки умственного недоразвития;
2) характерные особенности строения лица и черепа: выдающийся вперед высокий лоб, прогнатизм и удлиненные уши;
3) мальчики имеют увеличенные в размерах тестикулы (макроорхидизм).
Наблюдаются, кроме того, эпилептические припадки, синдром гиперактивности с дефицитом внимания, у более чем половины мальчиков аутизм и подобные аутизму расстройства, различные нарушения развития речи, персеверации, эхолалия, другие отклонения.
Женщины, унаследовавшие ломкую Х-хромосому с полной мутацией от своих матерей, могут быть склонны к развитию атипической депрессии, а также шизофреноподобного заболевания.
Синдром «кошачьего крика» — заболевание, обусловленное структурной аномалией 5-й пары хромосом (выпадение участка — делеция). Встречается преимущественно у девочек и характеризуется развитием умеренной или тяжелой умственной отсталости, задержкой физического развития и рядом диспластических признаков («антимонголоидный» разрез глаз, гипертелоризм, низкое расположение ушных раковин, поперечная складка ладоней и др.) Основным симптомом является своеобразный мяукающий тембр плача ребенка, связанный с аномалией строения гортани.
Синдром Вольфа—Хиршхорна.
В основе синдрома лежит изменение длины хромосомы из четвертой пары. Основные признаки заболевания у новорожденных: большое туловище, клювовидный нос и выступающее надпереносье, деформированные ушные раковины со складками, пучеглазие и колобома радужной оболочки (ее частичное отсутствие), общее недоразвитие во время беременности. Отмечается наличие четырех сгибательных складок на пальцах верхних конечностей.
Синдром Патау — комплекс врожденных пороков развития черепа, лица, нервной системы, органов слуха, зрения, внутренних органов. В основе заболевания лежит наличие добавочной хромосомы в 13-й паре. Синдром описан в 1960 г. американским педиатром Патау (К. Patau).
Клиническая картина характеризуется микроцефалией, расщелиной лица, двусторонним расщеплением верхней губы, полным расщеплением неба, маленькими глазными яблоками либо полным их отсутствием, короткой шеей, маленькими деформированными низко расположенными ушами, полидактилией, дистрофическими изменениями ногтей и костного скелета. Отмечаются также пороки развития сердца, желудка, кишечника и других органов.
Синдром трисомии-Х впервые описан в 1959 г. Частота данной патологии составляет среди новорожденных 0,1%, а среди умственно отсталых — 0,6%. Большинство лиц женского пола с трисомией-Х выявляется среди больных психиатрических лечебниц. Клиническая картина характеризуется аномалиями развития скелета, внутренних органов, различными психическими проявлениями и интеллектуальной недостаточностью. Среди полиморфизма признаков трисомии-Х наиболее характерными являются: низкий рост, аномалии ушей, прикуса, высокое стояние твердого неба, короткие пальцы, искривленный мизинец, широкий промежуток между 1 и 2 пальцами на стопах, синдактилия, недоразвитие половых функций.
Умственная отсталость проявляется в виде легкой или умеренной степени. Характерны эмоциональные расстройства (вспыльчивость, агрессивность, неустойчивость настроения и немотивированные поступки). Девочки с синдромом трисомии-Х с трудом, но в большинстве случаев (легкая степень умственной отсталости) обучаются в массовых школах.
Синдром Эдвардса — наследственное заболевание, обусловленное, как правило, трисомией 18-й хромосомы и проявляющееся множественными пороками развития органов и систем. Синдром описан в 1960 г. американским педиатром Эдвардсом (J. Edwards).
Клиническая картина заболевания характеризуется задержкой психического развития, множественными аномалиями лица, костно-мышечной системы, черепа и головного мозга.
К хромосомным синдромам, помимо вышеописанных, относится большая группа так называемых семейных форм умственной отсталости, когда совершенно точно доказано наличие данной патологии у близких родственников.
Синдром Аперта (акроцефалосиндактилия) — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, экзофтальмом, деформацией зубов и синдактилиями. Синдром описан французским педиатром Апертом (Е. Apert) в 1906 г.
Синдром Крузона — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, преждевременным срастанием швов черепа, уменьшением мозгового вещества, экзофтальмом, вторичной атрофией зрительных нервов, прямоугольным расположением большого пальца к кисти. Впервые синдром описан французским врачом Крузоном (О. Crouson) в 1912 г.
Синдром Сьегрена—Ларссона — наследственное заболевание, которое сопровождается умственной отсталостью, парезами (снижением силы) конечностей и ухудшением зрения.
Синдром Берьесона—Форсмана—Лемана — синдром характеризующийся умственной отсталостью в сочетании с избыточным весом. Впервые описан американскими врачами Берьесоном (М. Berjeson) Форсманом (Н. Foreman) и Леманом (О. Lehman) в 1963 г. Клиническая картина заболевания проявляется выраженным ожирением и прогрессирующей умственной отсталостью. Ожирение носит не равномерный характер. Жир откладывается преимущественно на бедрах, груди и лице, что придает своеобразный вид такому больному (бочкообразная карликовая фигура с заплывшим лицом, большими ушами и узкими разрезами глаз). У больных часто отмечаются эпилептические припадки. Умственная отсталость колеблется от умеренной до тяжелой степени. Данная патология встречается только у лиц мужского пола, но носителями патологического гена являются женщины.
Синдром Прадера—Вилли — наследственное заболевание, характеризующееся глубокой умственной отсталостью, низким ростом, гипогенитализмом, ожирением, резко выраженной мышечной гипотонией.
Синдром Книппеля—Фейля (синдром короткой шеи) — наследственное семейное заболевание, обусловленное врожденными аномалиями развития скелета и внутренних органов в сочетании с тяжелой степенью умственной отсталости. Клиника синдрома подробно описана французскими врачами Клиппелем Фейлем в 1912 г.
Аномалия развития характеризуется следующими проявлениями: короткой шеей как результат количественного уменьшения шейных позвонков, ограничением подвижности головы, расщеплением твердого неба, бочкообразной грудной клеткой, врожденными пороками сердца, добавочными долями или отсутствием отдельных долей легких, синдактилиями (сращение пальцев конечностей), глухотой вследствие заращения наружных слуховых проходов, сужением анального отверстия и многими другими симптомами. Интеллектуальная недостаточность является результатом тяжелой умственной отсталости
Лечение ЗПРР при хромосомных заболеваниях.
Основой лечения является уникальная методика патогенетической терапии речевых расстройств при хромосомной патологии — биофизическая активация нейромоторных структур, основу которого составляет щадящая стимуляция проводников нервной системы микротоками с использованием нейрофизиологического прибора. Метод лечения базируется как на активации самих речевых центров, так и на восстановлении нарушенных связей между центрами и полушариями головного мозга. Помимо этого, восстанавливаются разрозненные связи речевых центров с другими областями мозга, участвующими в реализации речевой функции. В процессе лечения формируется физиологичное, последовательное взаимодействие всех зон мозга, связанных с речепродукцией. В результате появляется речь.
Проведение биофизической активации сочетается с дополнительными методиками лечения, такими как — лимфомежклеточная терапия, которая применяется для регулирования интегративной деятельности и восполнения дефицита энергетической системы мозга и позволяющая применять малые дозы церебропротекторов, которые вводятся эндолимфатически и попадают в ткани головного мозга, минуя гематоэнцефалический барьер.
В качестве другого способа использования препаратов с нейротрофическим и антиоксидантным действием применяется методика эндоназального электрофореза кортексина, что позволяет вводить лекарственные препараты непосредственно в ткани головного мозга.
Исследования последних десятилетий выявили, что у большинства детей с речевыми и поведенческими проблемами в различной степени нарушены функции мозжечка и базальных ганглиев. Именно функционирование мозжечка определяет успешность ребенка в обучении. С этой целью применяется уникальная разработка Центра авиакосмической медицины — подошвенный имитатор опорной нагрузки «Корвит», применяемый для нейрофизиологической регуляции стато-кинетической функции ЦНС. В основе терапевтического воздействия аппарата «Корвит» лежит процесс активации опорной афферентации, отвечающей за нормализацию процессов возбуждения и торможения в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в головном мозге, способствующих восстановлению координации движений, и, опосредованно, улучшению речи и мышления.
Также для успешного лечения различных форм ЗПРР специалистами применяется одно из достижений современной науки — метод аудиовокальной терапии RUSTOMATIS. Прибор использует звукозаписи высокочастотных и низкочастотных компонентов. При чередовании такой музыки путем напряжения и расслабления у ребенка тренируется аппарат среднего уха – молоточек и стремечко, с помощью чего расширяется диапазон восприятия внешних факторов, увеличивается концентрация внимания, в мозг поступает новая информация и, как следствие исчезают многие нарушения и расстройства.
Обязательным звеном в лечебном комплексе у детей с наличием речевых расстройств является занятия с клиническим психологом, а также логопедическая коррекция, которая включает диагностику степени нарушений, ежедневные занятия, направленные на улучшение речевой функции и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
На фоне сочетания проведения биофизической активации со вспомогательными методиками лечения наблюдаются положительные изменения, которые могут быть видны уже через несколько процедур, но максимальный эффект развивается через полтора-три месяца после курса. Как правило, для закрепления полученных результатов и дальнейшего развития двигательных и когнитивных навыков специалистами центра рекомендуется повторный курс лечения через 5-6 месяцев.
Синдром Лежена — Генетика
Синдром Лежена это наследственное генетическое заболевание, связанное с изменением строения 5-ой хромосомы. Болезнь получила название по имени французского ученого Жерома Лежена в 1963 году. Для этого заболевания характерен необычный детский крик, напоминающий кошачий
Причиной возникновения синдрома Лежена является отсутствие фрагмента 5-ой хромосомы.
Симптомы
— Плач ребенка, напоминающий кошачье мяуканье
— Изменение в строении гортани или ее недоразвитие
— Отставание в развитии ребенка физическом и умственном
— Небольшой вес при рождении
— Пониженный мышечный тонус
— Лунообразное строение лица с широко расположенными глазами
— Врожденные пороки сердца
— Микроцефалия (уменьшенные размеры черепа и головного мозга при норме других частей тела)
— Характерно низкое расположение и деформация ушных раковин
— Выступающие лобные бугры
— Увеличенный клиновидный нос
— Аномалии внутренних органов (сосудов и почек)
— Ярко выраженные сложности с дыханием
— Паховые грыжи
— Малый размер нижней челюсти
— Косоглазие, астигматизм
Отдельные признаки синдрома Лежена имеют возрастные особенности. Исчезает со временем кошачий плач, мышечная гипотония, лунообразное лицо, а вот микроцефалия, косой разрез глаз приобретают более выраженные черты. Усиливается отставание и в психомоторном развитии ребенка. Учащается свистящее шумное дыхание и обостряются заболевания верхних дыхательных путей
Диагностика
Диагноз ставят исходя из клинических признаков и издающегося «крика котёнка». А лабораторные исследования подтверждают наличие синдрома при потери участка хромосомы 5
Лечение
Лечение синдрома направлено на устранение сопровождающихся симптомов. В первые месяцы жизни следует беречь ребенка от инфекционных заболеваний. Необходимо постоянное наблюдение со стороны педиатра и психоневролога. Врачами рекомендуются средства, стимулирующие психомоторное развитие ребенка, гимнастика и лечебный массаж
Профилактика
Своевременное проведение медико-генетического консультирования в семьях, где уже были дети с синдромом Лежена и проведение исследований на определение кариотипа родителей.
симптомы, диагностика и возможное лечение
Синдром кошачьего крика – редкая врожденная проблема, формирующаяся вследствие возникновения мутации в коротком плече пятой хромосомы. Частота встречаемости заболевания не превышает одного случая на 45 тыс. новорожденных. Симптомы включают в себя характерный плач, возникающий из-за деформации гортани, изменение строения черепа, отставание в развитии. Синдром Лежена, названный в честь генетика, впервые описавшего его в 1963 году, зачастую сочетается с другими врожденными патологиями. Лечение носит симптоматический характер и направлено на предупреждение развития осложнений. Важную роль играют также занятия с дефектологом и устранение речевых нарушений.
Причины возникновения синдрома кошачьего крика
Заболевание является врожденным и характеризуется способностью к наследованию. Это означает, что патология имеет генетическую природу. Синдром Лежена формируется в результате мутации, приводящей к изменению строения 5-й хромосомы, а именно ее короткого плеча. Эта структура либо укорачивается, либо теряется полностью. Встречается и мозаичная форма болезни. Среди причин возникновения синдрома кошачьего крика выделяют воздействие следующих неблагоприятных факторов:
- Значение имеет возраст будущей матери. По мере старения женского организма повышается риск развития генетических мутаций, способных приводить к формированию врожденных заболеваний. Данная закономерность прослеживается у представительниц прекрасного пола старше 45 лет.
- Употребление алкоголя и курение в подростковом возрасте и во время беременности. Вредные привычки оказывают негативное влияние как на организм матери, так и на плод.
- Заражение вирусными и бактериальными инфекциями в период вынашивания ребенка. Ряд болезнетворных агентов способны изменять процесс развития малыша, что приводит к формированию различных нарушений.
- Воздействие радиации – один из ключевых факторов в возникновении спонтанных мутаций. Излучение способно незаметно проникать в клетки человеческого организма и негативно сказываться на структуре генетического материала.
Характерные симптомы патологии
В медицине принято выделять несколько основных клинических признаков синдрома кошачьего крика:
- Характерный плач новорожденного. Этот симптом регистрируется сразу после появления ребенка на свет. Издаваемые звуки напоминают мяуканье, из-за чего заболевание и получило свое название. Появление данного признака связано с изменением структуры надгортанника, хрящей и слизистой оболочки в области глотки. Синдром мяуканья исчезает к годовалому возрасту.
- Деформации черепа. Наиболее распространенной является микроцефалия, то есть уменьшение размеров головы ребенка. Подобное нарушение провоцирует развитие неврологической симптоматики в будущем, а также является органической основой умственной отсталости.
- Изменение расположения и разреза глаз. Такое проявления связано с деформацией структуры черепа. Существует несколько вариаций данного клинического признака. У некоторых детей встречается антимонголоидный вариант нарушения, другие страдают от гипертелоризма – увеличения расстояния между глазами (фото представлено ниже).
- Малыши с синдромом Лежена рождаются со сниженной массой тела и мышечной слабостью. В дальнейшем они значительно отстают от сверстников в развитии как в физическом, так и в умственном.
- Зачастую у малышей недоразвита нижняя челюсть. Это не только приводит к возникновению характерных черт лица, но и усугубляет недоразвитость ребенка, поскольку значительно затрудняет процесс грудного вскармливания.
Сопутствующие заболевания
Особенность врожденных патологий в том, что они зачастую сочетаются между собой. При синдроме Лежена, кроме характерных изменений структуры черепа и отставания в развитии, диагностируются и другие поражения. У детей с заболеванием высок риск возникновения пороков сердца, значительно ухудшающих прогноз недуга. Нарушается также работа желудочно-кишечного тракта, что сопровождается частыми запорами. Малыши страдают от дефектов костно-мышечного аппарата, например, изменения строения кистей рук и искривления позвоночника. Зачастую страдают также почки и печень, формируются тяжелые нарушения метаболизма. Распространенной проблемой при синдроме кошачьего крика являются грыжи, требующие хирургического лечения.
Диагностика
Подтверждение наличия заболевания у новорожденного проводится как на основании клинических признаков, так и при помощи специальных тестов. Деформации черепа, характерный плач и отставание в развитии дают основание предположить проблему. Основу диагностики синдрома кошачьего крика составляет определение кариотипа как малыша, так и родителей. Для подтверждения наличия отклонения в пренатальном периоде делается УЗИ.
В ходе исследования выявляются признаки хромосомных делеций: изменение количества околоплодных вод, деформация костей черепа, пороки сердца. Однако на этом этапе поставить точный диагноз не удается. После рождения ребенка, кроме генетических анализов, используется ЭХО, рентген, а также гематологические тесты, позволяющие оценить функцию внутренних органов.
Пренатальная диагностика не слишком распространена при выявлении заболевания. Например, изучение структуры хромосом плода практически не используется. При этом в ряде случаев проводится исследование ребенка при помощи ультразвука. Косвенными показателями, указывающими на формирование патологии, являются два признака: не иммунная водянка плода, а также вентрикуломегалия, то есть увеличение объемов мозговых желудочков.
К числу редких симптомов заболевания в антенатальном периоде относят формирование кист сосудистых сплетений плаценты, хотя и это проявление не является специфическим. Есть также данные об обнаружении в ходе УЗИ микроцефалии и гипоплазии мозжечка.
Несмотря на то что генетический анализ является самым достоверным методом при постановке диагноза, как в пренатальный период, так и после рождения ребенка, подобные тесты используются довольно редко. Это связано с тем, что отклонение в строении 5-й хромосомы не всегда сопровождается возникновением синдрома кошачьего крика.
Подобные исследования проводятся врачами только в тех случаях, когда у пациента отмечается наличие специфических клинических признаков, позволяющих заподозрить данную проблему. В таких случаях осуществление генетического анализа используется с целью дифференциации заболевания от других дефектов со сходной симптоматикой.
Существующие методы лечения
Специфических мер борьбы с синдромом кошачьего крика не разработано. Если у ребенка выявляются сопутствующие заболевания, например пороки клапанного аппарата сердца, проводится их оперативная коррекция. К хирургическим техникам прибегают также при наличии у малыша врожденных грыж. В ряде случаев, когда пациенты страдают от недостаточности функции почек и печени, проводится терапия в зависимости от стадии процесса. Лечение при заболевании паллиативное, то есть носит симптоматический характер, а также направлено на профилактику осложнений.
Важным этапом терапии является стимуляция нормального умственного и физического развития ребенка. Для этого применяется физиотерапия, основанная на использовании воздействия тепла, и занятия в бассейне. Хорошие результаты показывают также массажные техники и лечебная физкультура, направленная на улучшение тонуса мышц. Детям требуется психотерапия, позволяющая скорректировать отклонения в функции высшей нервной деятельности.
В ряде случаев пациентам рекомендуется медикаментозная терапия препаратами из группы фенобарбиталов или бензодиазепинов. Занятия с логопедом необходимы, особенно в случаях деформации челюсти.
Даже при адекватном лечении дети с синдромом Лежена не могут обслуживать себя. Они нуждаются в постоянном надзоре и уходе, в том числе и в случаях, когда пациенты доживают до преклонного возраста.
Прогноз и профилактика
Исход заболевания определяется выраженностью изменений, а также наличием сопутствующих патологий. У пациентов повышен риск развития осложнений со стороны дыхательной и мочевыделительной систем, а также имеются значительные трудности в сфере социальных взаимодействий. В целом прогноз при синдроме Лежена стремится к благоприятному. Благодаря современным методам диагностики и лечения поражений различных органов, больным удается дожить до 40–50 лет.
Профилактика патологии сводится к правильному планированию и ведению беременности. Будущим родителям, семейный анамнез которых осложнен случаями выявления хромосомных мутаций, рекомендуется проходить генетические тесты. Женщинам в период вынашивания ребенка следует избегать воздействия стрессов, правильно питаться, а также отказаться от вредных привычек. Специфических методов профилактики проблемы не разработано.
Отзывы
Юлия, 28 лет, г. Москва
Сын родился с синдромом кошачьего крика. Это такая генетическая болезнь, при которой дети отстают в развитии и у них изменяется строение черепа. Хорошо, что никаких других заболеваний у малыша не обнаружили. Сейчас сын занимается с логопедом. Мы ходим на сеансы массажа, а еще делаем специальную гимнастику. Болезнь не лечится, но терапия помогает ребенку чувствовать себя лучше.
Арсений, 35 лет, г. Воронеж
Когда дочь родилась, сразу стало ясно, что у нее какая-то генетическая патология. У ребенка была маленькая нижняя челюсть, широко посаженные глаза, и плакала она, как будто мяукала. Оказалось, это синдром кошачьего крика. Врачи сказали, что болезнь не лечится, но рекомендовали физиопроцедуры. Сейчас дочка занимается с дефектологом и делает большие успехи.
Загрузка…
Синдром кошачьего крика (Лежена): причины, диагноз, прогноз
Синдром кошачьего крика (СКК) — генетическое заболевание, обусловленное структурным дефектом пятой хромосомы: отсутствием ее небольшой части. При этом общее число носителей наследственной информации остается нормальным. Врачи диагностируют болезнь с первого дня жизни новорожденного.
Свое наименование недуг получил из-за характерного, пронзительного и высокотонального крика ребенка, напоминающего кошачье мяуканье. Данное клиническое проявление связано с патологическим развитием гортани, мягкостью хрящевой ткани и сужением надгортанника. СКК проявляется признаками врожденных патологий внутренних органов, характерными внешними данными, зрительными расстройствами, выраженным нарушением интеллекта.
мальчик с синдромом кошачьего крика в 8 месяцев, 2 года, 4 и 9 лет
примеры детей с выраженными дефектами
Недуг имеет еще одно название – синдром Лежена, в честь генетика из Франции, впервые описавшего эту хромосомную болезнь в 1963 году. Ее распространенность составляет 1 случай на 60 тысяч новорожденных и не зависит от национальности, климата, социальных и производственных факторов, а также региона проживания. У девочек болезнь обнаруживается чаще, чем у мальчиков. Известно, что хромосомные нарушения не лечатся, а больные дети с рождения являются инвалидами. Такие малыши не могут сами справиться с проявлениями синдрома и полноценно жить.
Диагноз синдрома кошачьего крика подтверждается результатами кариологического анализа крови с применением одного из методов идентификации хромосом. Специфического этиотропного лечения СКК не существует. Обычно проводится симптоматическая терапия с применением препаратов, стимулирующих психомоторное развитие, массажа и ЛФК. Больные дети нуждаются в хирургической коррекции тяжелых врожденных аномалий. Они часто страдают и погибают от серьезных осложнений.
Прогноз при СКК относительно неблагоприятный по сравнению с другими хромосомными аномалиями. Больные дети могут дожить до зрелых лет, если им будет обеспечен хороший уход, забота и лечение. При этом их психологическое, физическое развитие и интеллект всегда останутся неполноценными.
Этиопатогенетические факторы
Синдром кошачьего крика развивается у лиц, имеющих структурную деформацию пятой хромосомы. Данное генетическая патология обусловлена утратой части или целой половины короткого плеча. У хромосомы может полностью отсутствовать плечо – небольшой фрагмент с множеством генов. Особенности клинической картины синдрома связаны не с размером ее потерянного участка, а с определенным фрагментом. Клетки с поврежденными хромосомами не могут полноценно синтезировать химические соединения. Они производят дефектные белки, что и приводит к появлению характерных симптомов болезни.
Возможна транслокация части плеча 5 хромосомы на иную, негомологичную хромосому. Подобные хромосомные аберрации обычно вызваны спонтанной мутацией генов и лишь в 10% случаев они наследуются от родителей.
СКК может развиться при любом из этих вариантов мутационных изменений:
- Полное отсутствие короткого плеча — самый распространенный вариант патологии, приводящий к тяжелым врожденным дефектам внутренних органов.
- Потеря части плеча хромосомы сопровождается развитием меньшего числа аномалий.
- Кольцевая хромосома – замкнутая двухцепочечная молекула ДНК, у которой оба конца укорочены и сцеплены друг с другом. В результате подобной трансформации теряется небольшой конечный участок хромосомы. Если делеция затрагивает ключевые гены, развивается синдром Лежена.
- Мозаицизм – легкий вариант мутации, встречающийся крайне редко. Ребенок получает от родителей нормальные хромосомы. Их структурные изменения происходят после образования зиготы. При этом большинство клеток имеет нормальный геном, а их меньшая часть – набор генов, характерный для СКК. У больных детей имеется меньше врожденных аномалий: их физическое развитие остается нормальным, внутренние органы функционируют полноценно, но имеется выраженное снижение интеллекта.
Причины образования структурного дефекта пятой хромосомы в настоящее время до конца не определены. Считается, что их огромное количество, и ни одна из них не является главной. На развитие патологии оказывает влияние совокупность факторов внешней среды, которые повреждают яйцеклетки и сперматозоиды или нарушают процесс деления зиготы.
Факторы, провоцирующие развитие синдрома:
- возраст матери,
- вредные привычки,
- наркотики,
- влияние медикаментов,
- инфекционные заболевания беременных — цитомегаловирусы, герпес-вирусы,
- ионизирующее излучение,
- неблагоприятные условия среды,
- воздействие химикатов и иных токсинов,
- оперативные вмешательства при беременности.
Имеет место генетический фактор, когда патологии кариотипа наблюдаются у родителей. Если в семье имеется ребенок с СКК, то вероятность повторного рождения малыша с аналогичной аномалией повышается.
Симптоматика
Новорожденные с СКК появляются на свет с пренатальной гипотрофией – недостаточной массой тела. Их внутренние органы и системы не до конца сформированы. При этом рождаются такие дети чаще всего в положенный срок. Беременность протекает хорошо, патологических процессов при ней не наблюдается.
внешние признаки синдрома кошачьего крика
Клиника синдрома кошачьего крика:
- Характерный плач ребенка обусловлен дефектом гортани, который формируется внутриутробно. Специфический крик на высоких тонах напоминает кошачье мяуканье. У больных детей размеры надгортанника намного меньше, чем у здоровых, дыхательные пути сужены, хрящевая ткань более мягкая, имеются складки на слизистой оболочке гортани. Изменение тембра голоса — основной симптом патологии, который присущ каждому больному. Это патономоничный признак, который исчезает к концу первого или второго года жизни.
- У 85% новорожденных наблюдается микроцефалия. Маленькая голова новорожденных вытягивается в продольном направлении. Краниометрия подтверждает наличие данной аномалии. У больных с микроцефалией в будущем прогрессирует умственная отсталость.
- У некоторых новорожденных встречается антимонголоидный разрез глаз. Узкие глазные щели имеют опущенные внешние и приподнятые внутренние уголки. Больные с СКК страдают страбизмом, астигматизмом, снижением остроты зрения, катарактой, близорукостью. Для новорожденных с СКК характерно наличие гипертелоризма, депигментированной сетчатки и эпикантуса.
- Дефекты слухового анализатора проявляются низким расположением ушных раковин, патологически мягкой хрящевой тканью, образованием небольших плотных узелков вокруг ушей. У больных орган слуха имеет маленький размер и суженный слуховой проход.
- Микрогения или микрогнатия – гипоплазия костей нижней челюсти с втянутостью подбородка. При двусторонней микрогнатии уменьшенная нижняя челюсть остается симметричной, а между зубами образуются широкие промежутки. При односторонней микрогнатии недоразвита только одна из ветвей нижней челюсти, что делает лицо несимметричным. Данный признак нарушает нормальный процесс грудного вскармливания. Сосательный рефлекс быстро угасает.
- Низкая масса тела характерная практически для всех новорожденных с СКК. В среднем их вес при рождении не превышает 2500 грамм.
- Синдактилия — неполное или полное сращение двух или более пальцев кисти или стопы. Обычно они соединены кожной перепонкой, которую легко рассекают во время операции. Если сращена костная ткань, исправить ситуацию намного сложнее. Клинодактилия – искривление пальцев или искажение их положения относительно оси конечности, не позволяющее полностью согнуть или разогнуть кисть.
- Косолапость – порок развития костно-суставного аппарата, при котором стопа сильно отклоняется внутрь по отношению к голени. Дети, имеющие такой дефект, поздно начинают ходить. Поражение костно-мышечной системы проявляется вывихом бедра, развитием сколиоза, плоскостопия, паховыми или пупочными грыжами.
- Патология дыхательной системы у больных детей проявляется частыми простудными заболеваниями, высоким риском развития пневмоний, которые нередко заканчиваются летальным исходом.
- Врожденные пороки сердца и сосудов – открытый артериальный поток, ДМПП, ДМПЖ, тетрада Фалло, количественное и структурное изменение сосудов, приводящее к дисциркуляторным нарушениям и развитию сердечной недостаточности. У детей синеет кожа, возникает слабость, одышка, усиливается сердцебиение, колеблется давление, возможны обмороки. Врожденные пороки сердца — основная причина летального исхода при СКК.
- Аномалии почек – гидронефроз, атрофия почечной ткани, подковообразная форма почек.
- Гипоспадия – патологическое развитие полового члена, когда внешнее отверстие уретры открывается не на головке полового органа, а на его стволе. Больные дети испытывают трудности при мочеиспускании, которое становится болезненным или полностью невозможным.
- Врожденные дефекты пищеварительного тракта — сужение или полное закрытие просвета пищеварительной трубки на всем ее протяжении. У больных нарушается процесс глотания и переваривания пищи, возникает задержка кала, рвота, пропадает аппетит, больные резко худеют. Нарушение нервной регуляции кишечника проявляется запорами.
- К прочим признакам синдрома относятся: готическое небо, нарушение глотания, гиперсаливация, расщепление язычка, плоская спинка носа, расщелина неба, шумное дыхание, цианоз, аномальный прикус.
Дети с СКК отстают в умственном и физическом развитии от своих сверстников. У них чаще развиваются офтальмологические патологии, снижается тонус мышц, нарушается координация движений, часто возникают перепады настроения: плач беспричинно сменяется истеричным хохотом. В коллективе дети агрессивны и чрезмерно активны. У больных развивается имбецильность или олигофрения. У них появляются проблемы с речью, снижается способность к обучению. Больные дети не могут освоить обычную школьную программу, учатся на дому или в спецшколах.
У больных лицевые кости намного шире черепных, что придает лицу характерную форму — лунообразную. Нарушение осанки и короткая шея также являются типичными признаками синдрома. Дискоординация движений, неустойчивая походка, частые падения обусловлены гипоплазией мозжечка.
Дети с синдромом кошачьего крика рано погибают от врожденных пороков внутренних органов. Лишь 10% больных умирает в возрасте 12-15 лет, и только единицы переживают 50-летний рубеж. У лиц с СКК репродуктивная функция полностью сохраняется, несмотря на развитие у женщин двурогой матки, а у мужчин – гипотрофии или атрофии яичек.
Диагностика
Чтобы правильно диагностировать любое заболевание, в том числе хромосомное, необходимо тщательно обследовать больного. Специалисты собирают семейный или наследственный анамнез, проводят кариотипирование супругов, пренатальный скрининг и инвазивные процедуры.
В центрах пренатальной диагностики выявляют женщин с высоким риском появления на свет детей с врожденными аномалиями. С помощью целого ряда диагностических мероприятий, проводимых на дородовом этапе, можно выявить различные хромосомные аномалии плода в самом начале беременности.
Методы пренатальной диагностики:
- Кариотипирование родителей – выделение клеток, изучение их ядра, окрашивание материала и микроскопическое изучение хромосом. Нормальный женский кариотип — 46 ХХ, мужской — 46 ХУ. При наличии каких-либо отклонений у родителей шансы родить больного ребенка возрастают во много раз.
- УЗИ-признаки генетических патологий плода – увеличение воротниковой зоны, недостаток или избыток околоплодных вод, ВПС, короткоголовость, непроходимость кишечника. При наличии данных признаков плод с СКК погибает до рождения.
- В крови у беременных определяют количество хорионического гонадотропина, протеина А, эстриола, альфа-фетопротеина.
делеция небольшого участка хромосомы у ребенка
Кордоцентез — исследование пупочной крови плода, позволяющее на 100% подтвердить или исключить заболевания, при которых изменяется количество или структура хромосом.
- Исследование околоплодных вод на хромосомные и генетические заболевания плода начинается с пункции амниотической оболочки, забора материала и его последующего исследования.
- Хорионбиопсия — метод пренатальной диагностики, заключающийся в получении ворсин хориона и исследовании биоматериала. Проводят этот пренатальный тест с 10 по 12 неделю беременности.
Все инвазивные методики проводят специальными тонкими иглами под контролем УЗИ. Данные анализы показаны женщинам в возрасте старше 30 лет или имеющим в семейном анамнезе хромосомные заболевания.
Послеродовая диагностика:
- Всех новорожденных с признаками СКК сразу после рождения обследуют неонатологи и врачи-педиатры.
- Данные кардиограммы и эхокардиографического исследования позволяют выявить врожденные пороки сердца.
- Рентгенографическое или ультразвуковое исследование необходимо для обнаружения патологий ЖКТ, почек.
- Чтобы диагностировать заболевание, у новорожденного берут мочу и кровь на анализы.
- Проводят цитогенетическое исследование хромосомного набора пациента.
- Новорожденным показана консультация специалистов в области детской кардиологии, офтальмологии, урологии, ортопедии и других узких областях медицины.
Лечебные мероприятия
Синдром Лежена — редкая наследственная болезнь, которая в настоящее время все еще остается неизлечимой. Больным показана симптоматическая терапия, общеукрепляющий и тонизирующий массаж, физическая культура, физиотерапевтические процедуры, логопедическая помощь, занятия с дефектологами, сеансы психотерапии.
При наличии врожденных пороков сердца, почек или других внутренних органов больным требуется хирургическое вмешательств. Таких пациентов направляют на консультацию к хирургам, которые проведут дополнительное обследование и сделают операцию.
Профилактические мероприятия синдрома:
- Полное обследование лиц, желающих стать родителями,
- Исключение негативного воздействия факторов внешней среды на организм беременной женщины,
- Прохождение цитогенетического обследования для исключения носительства патологии.
Прогноз патологии неблагоприятный. Больные дети практически не доживают до 10 лет. Обычно они умирают в первые годы жизни от сопутствующих недугов — пороков сердца, почек или других органов. Медицине известны единичные случаи, когда пациенты с СКК умирали после 40−50 лет. Продолжительность жизни больных определяется степенью повреждения хромосомы, уровнем ухода, образом жизни, своевременностью и качеством медицинской помощи. При правильном лечении и адекватном обучении дети могут научиться писать, читать и выполнять простые задания.
СКК – редкое, но опасное заболевание, которое лучше предотвратить, поскольку вылечить его невозможно. Беременные женщины должны заботиться о себе и своем будущем ребенке, не пить, не курить, правильно питаться, вести здоровый образ жизни. Если у ребенка были обнаружены врожденные аномалиями, ему требуется особый уход, забота и повышенное внимание.
Видео: ТВ-шоу о синдроме кошачьего крика
Синдром Кри дю Чата — NORD (Национальная организация по редким заболеваниям)
УЧЕБНИКИ
Mezoff AG. Синдром Кри-дю-Шат. Руководство NORD по редким заболеваниям. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003: 175.
Rimoin D, Connor JM, Pyeritz RP, Korf BR. Ред. Принципы и практика медицинской генетики Эмори и Римоан. 4-е изд. Черчилль Ливингстон. Нью-Йорк, штат Нью-Йорк; 2002: 1209-10.
Горлин Р.Дж., Коэн MMJr, Hennekam RCM. Ред. Синдромы головы и шеи.4-е изд. Издательство Оксфордского университета, Нью-Йорк, штат Нью-Йорк; 2001: 51.
Джонс KL. Эд. Распознаваемые модели пороков развития человека Смита. 5-е изд. W. B. Saunders Co., Филадельфия, Пенсильвания; 1997: 44.
СТАТЬИ ИЗ ЖУРНАЛА
Уллах И., Махаджан Л., Магнусон Д. Недавно признанная ассоциация болезни Гиршпрунга с синдромом Кри-дю-чат. Am J Gastroenterol. 2017; 112: 185-186.
Nguyen JM, Qualmann KJ, Okashah R, Reilly A, Alexeyev MF, Campbell DJ. Удаление 5p: текущие знания и будущие направления.Am J Med Genet C Semin Med Genet. 2015 сентябрь; 169 (3): 224-38.
Родригес-Кабальеро А., Торрес-Лагарес Д., Родригес-Перес А., Серрера-Фигалло М. А., Эрнандес-Гисадо Дж. М., Мачука-Портильо Г. Синдром кри-дю-чата: критический обзор. Oral Patol Oral Cir Bucal. 2010; 15: e473-8.
Hill C, Moller JH, Finkelstein M, Lohr J, Schimmenti L. Синдром Кри-дю-чата и врожденный порок сердца: обзор ранее зарегистрированных случаев и представление еще 21 случая от консорциума педиатрической кардиологической помощи.Педиатрия. 2006; 117: 924-7.
Laczmanska I, Stembalska A, Gil J, Czemarmazowicz H, Sasiadek M. Cri du chat синдром, определяемый диагностическими проблемами делеции 5p15.3? Pter. Eur J Med Genet. 2006; 49: 87-92.
Mainardi PC, Pastore G, Castronovo C, et al., Естественная история критического синдрома. Отчет Итальянского Регистра. Eur J Med Genet. 2006; 49: 363-83.-9.
Кондо Т., ШимоКава О., Харада Н., Дои Т. и др., Корреляция генотип-фенотип 5p-синдрома: ловушка диагностики.J Hum Genet. 2005; 50: 26.
Посмык Р, Панасюк Б, Яценко С.А., Станкевич П, Мидро АТ. Естественная история ребенка с синдромом моносомии 5 (синдром кошачьего крика / кри-дю-чат) в течение 18 лет наблюдения. Genet Couns. 2005; 16: 17-25.
Чжан Х, Снайдерс А., Сегрейвс Р. и др., Картирование с высоким разрешением взаимосвязей генотип-фенотип при критическом синдроме с использованием сравнительной геномной гибридизации массива. Am J Hum Genet. 2005; 76: 312-6.
Van Buggenhout GJ, Pijkels E, Holvoet M, et al., Синдром Кри-дю-чат: изменение фенотипа у пожилых пациентов. Am J Med Genet. 2000; 90: 203-15.
ИНТЕРНЕТ
Синдром Cerruti Mainardi P. Cri du Chat. Энциклопедия Orphanet, 5 сентября 2006 г. Доступно по адресу: http://www.ojrd.com/content/1/1/33, дата обращения 5 сентября 2017 г.
Mithilesh, K. Cri-du-Chat Syndrome. Emedicine Journal, 30 сентября 2005 г. Последнее обновление: 09.06.2017. Доступно по адресу: http://www.emedicine.com/ped/topic504.htm Доступно 5 сентября 2017 г.
McKusick VA., Ed. Интернет-Менделирующее наследование в человеке (OMIM).Балтимор. MD: Университет Джона Хопкинса; Запись №: 123450; Последнее обновление: 01.08.2017. Доступно по адресу: http://omim.org/entry/123450 (дата обращения: 5 сентября 2017 г.).
Кариотип кри-дю-чата или кошачьего плача, синдром, также известный как синдром, известный как синдром критипа. Фотография, картинки, изображения и сток-фотография без роялти. Image 154005479.
Кариотип кри-дю-чата, или кошачий крик, синдром, также известный как синдром кри-ду-чата Фотография, картинки, изображения и сток-фотография без роялти. Изображение 154005479.
Кариотип Cri du chat, или кошачьего плача, синдром, также известный как 5p-синдром.Милый кот плачет в чате ДНК на белом фоне. Кариотип хромосомы ДНК, кри (кри), также известный как кариотип-кри, представляет собой частичное воспроизведение хромосомы. Понятие генетики. Милый плачущий кот (кри) с удалением кариотипа. Мужской персонаж медицинской науки. Также известен как синдром крид-д. Болтаем с друзьями. Концепция медицины и генетики.
M
L
XL
Таблица размеров
| Размер изображения | Идеально подходит для |
| S | Интернет и блоги, социальные сети и мобильные приложения. |
| млн | Брошюры и каталоги, журналы и открытки. |
| л | Плакаты и баннеры для дома и улицы. |
| XL | Фоны, рекламные щиты и цифровые экраны. |
Используете это изображение на предмете перепродажи или шаблоне?
Распечатать
Электронный
Всесторонний
5000 x 4000 пикселей
|
42.3 см x
33,9 см |
300 точек на дюйм
|
JPG
Масштабирование до любого размера • EPS
5000 x 4000 пикселей
|
42,3 см x
33,9 см |
300 точек на дюйм
|
JPG
Скачать
Купить одно изображение
6 кредитов
Самая низкая цена
с планом подписки
- Попробуйте 1 месяц на 2209 pyб
- Загрузите 10 фотографий или векторов.
- Нет дневного лимита загрузок, неиспользованные загрузки переносятся на следующий месяц
221 ру
за изображение любой размер
Цена денег
Ключевые слова
Похожие изображения
Нужна помощь? Свяжитесь со своим персональным менеджером по работе с клиентами
@ +7 499 938-68-54
Мы используем файлы cookie, чтобы вам было удобнее работать.Используя наш веб-сайт, вы соглашаетесь на использование файлов cookie, как описано в нашей Политике использования файлов cookie
.
Принимать
Синдром Кри-дю-чат: основы практики, патофизиология, эпидемиология
Автор
Митилеш Кумар Лал, MD, MBBS, MRCP, FRCPCH, MRCPCH (Великобритания) Консультант неонатолог, клинический директор по неонатальным службам, младший медицинский директор (повторная валидация), Больница Университета Джеймса Кука, Доверительный фонд NHS South Tees Foundation; Британский председатель, экзамен по теории MRCPCH, старший клинический экзаменатор MRCPCH, Королевский колледж педиатрии и здоровья детей (Великобритания)
Митилеш Кумар Лал, MD, MBBS, MRCP, FRCPCH, MRCPCH (Великобритания) является членом следующих медицинских обществ: Педиатрическое общество, Общество педиатрических исследований, Британская ассоциация перинатальной медицины, Британская медицинская ассоциация, Неонатальное общество, Непальская медицинская ассоциация, Королевский колледж педиатрии и здоровья детей, Королевский колледж врачей
Раскрытие информации: раскрывать нечего.
Специальная редакционная коллегия
Мэри Л. Виндл, PharmD Адъюнкт-профессор фармацевтического колледжа Медицинского центра Университета Небраски; Главный редактор Medscape Drug Reference
Раскрытие информации: нечего раскрывать.
Главный редактор
Мария Декарт, доктор медицины Профессор, кафедра генетики человека и кафедра педиатрии, Университет Алабамы, Медицинская школа Бирмингема
Мария Декарт, доктор медицины, является членом следующих медицинских обществ: Американская академия педиатрии, Американский медицинский колледж Генетика и геномика, Американская медицинская ассоциация, Американское общество генетики человека, Общество наследственных метаболических нарушений, Международное общество скелетной дисплазии, Юго-восточная региональная генетическая группа
Раскрытие: нечего раскрывать.
Дополнительные участники
Гарольд Чен, доктор медицины, магистр медицины, FAAP, FACMG Профессор, кафедра педиатрии, Медицинский центр государственного университета Луизианы
Гарольд Чен, доктор медицины, магистр медицины, FAAP, FACMG является членом следующих медицинских обществ: Американская академия педиатрии, Американский колледж медицинской генетики и геномики, Американская медицинская ассоциация, Американское общество генетики человека
Раскрытие информации: нечего раскрывать.
Дэвид Флэннери, доктор медицины, FAAP, FACMG Заместитель председателя отдела образования, руководитель секции медицинской генетики, профессор, кафедра педиатрии, Медицинский колледж Джорджии
Дэвид Флэннери, доктор медицины, FAAP, FACMG является членом следующих медицинских организаций. общества: Американская академия педиатрии, Американский колледж медицинской генетики и геномики
Раскрытие информации: нечего раскрывать.
Синдром кри-дю-чата — «Крик кошки»
26 мая 2016
Синдром 5P минус, также называемый синдромом Кри дю Шат (или CdCS), является генетическим заболеванием, вызванным отсутствием части хромосомы номер 5. Те, кто родился с этой отсутствующей или укороченной хромосомой, имеют характерный «кошачий крик» или кошачий плач (тихий плач) из-за неразвитой гортани, который может усиливаться по мере взросления ребенка. Меньшая масса тела при рождении, меньшая окружность головы и задержка в развитии часто встречаются у детей с синдромом Кри дю Шат.
В течение мая Общество 5P Minus и Cri du Chat.org работают над информированием других об этом чрезвычайно редком заболевании, которым страдает примерно один из 50 000 новорожденных во всем мире.
Виртуальный 5K для 5P Минус
Чтобы привлечь внимание к программам, которые приносят пользу сообществу с синдромом Кри-дю-Чат, и привлечь деньги, сторонники могут запланировать виртуальную прогулку. Мероприятие может быть прогулкой, пробежкой, пикником или любым другим мероприятием. По всему миру создано более 20 «команд».Проверьте их, выполнив поиск # 5pminus5k в социальных сетях.
Кампания с полосатыми носками
Наглядный и забавный способ распространения информации — поощрение сторонников носить полосатые носки, один длинный и один короткий, чтобы представить всю 5-ю хромосому и удаленную 5-ю хромосому. Используйте хэштеги #criduchat, #stripysocks и #criduchatsyndromeawareness при публикации изображений в Twitter, Instagram и Facebook.
Талисман C5
Еще один изобретательный способ — 5P Minus Society и Cri du Chat.org подчеркивают осведомленность о CdCS во всем мире, прося сторонников загрузить C5, характерную иллюстрацию хромосомы C5 с укороченным плечом. Вы можете скачать C5, брать его с собой куда угодно, сделать снимок (аналогично проекту Flat Stanley) и использовать хэштеги выше или # SeeC5. Учителям, семьям и сторонникам рекомендуется загрузить C5 в виде раскраски.
Факты о синдроме Кри дю Чат
- Синдром Кри-дю-Шат, также известный как 5P- (Five P Minus), возникает, когда происходит потеря генетического материала на 5 -й
- Его основная характеристика — это кошачий крик (тихий крик), который возникает из-за недоразвитой гортани.По мере роста ребенка и укрепления гортани звук немного углубляется.
- Другие характеристики могут включать низкий вес при рождении, небольшую окружность головы, небольшой подбородок, низко посаженные уши, кожные складки во внутренних уголках глаз, широкий нос, низкий мышечный тонус, задержку когнитивных функций и задержку выразительной речи.
- Это редкое генетическое заболевание поражает 1: 50 000 рождений. Большинству людей диагноз ставят при рождении, потому что поставщики замечают основные характеристики, но некоторым людям диагноз ставят позже из-за задержек физического и когнитивного развития.
- Синдром Кри дю Шат был открыт в (1963) доктором Жеромом Леженом во Франции.
Для получения дополнительной информации посетите http://www.fivepminus.org или http://www.criduchat.org.
О компании Epic Health Services
Если вашему ребенку или близкому человеку требуется помощь, мы в Epic Health Services предлагаем индивидуальный уход на дому и терапевтические услуги, которые отвечают потребностям наших пациентов, а также улучшают качество их жизни. Для получения более подробной информации свяжитесь с нами.
Источники:
http://www.fivepminus.org/Awareness
http://www.criduchat.org
| От 80% до 99% людей имеют эти симптомы | ||
| Кошачий крик | кошачий крик | 0200046 |
| Эпикантус | Складки глаз Выраженные складки глаз [ более ] | 0000286 |
| Высокий голос | 0001620 | |
| Умственная отсталость тяжелой степени | Ранняя и тяжелая умственная отсталость Умственная отсталость, тяжелая форма Тяжелая умственная отсталость [ более ] | 0010864 |
| Низко посаженные уши, повернутые назад | 0000368 | |
| Микроцефалия | Аномально маленький череп Уменьшение окружности черепа Уменьшенный размер черепа Уменьшенная окружность головы Небольшая окружность головы [ более ] | 0000252 |
| Микроретрогнатия | Маленький втянутый подбородок | 0000308 |
| Мышечная гипотония | Низкий или слабый мышечный тонус | 0001252 |
| Круглая грань | Круглое лицо Круглый внешний вид лица Круглая форма лица [ более ] | 0000311 |
| Сильная задержка глобального развития | 0011344 | |
| Широкая переносица | Широкая носовая перемычка Широкий корень носа Расширенный носовой мост Увеличенная ширина переносицы Увеличенная ширина переносицы Увеличенная ширина переносицы Увеличенная ширина переносицы Носовая перемычка широкая Широкая переносица Расширенный носовой мост [ более ] | 0000431 |
| 30% -79% людей имеют эти симптомы | ||
| Нисходящие глазные щели | Наклон отверстия между веками вниз | 0000494 |
| Высокое небо | Повышенное небо Увеличенная небная высота [ более ] | 0000218 |
| Гипертелоризм | Широко посаженные глаза Широко расставленные глаза [ более ] | 0000316 |
| Задержка внутриутробного развития | Дефицит дородового роста Задержка внутриутробного развития [ более ] | 0001511 |
| Сколиоз | 0002650 | |
| Короткая шея | Уменьшенная длина шеи | 0000470 |
| Низкий рост | Уменьшенный рост Маленький рост [ более ] | 0004322 |
| Маленькая рука | Непропорционально маленькие руки | 0200055 |
| 5% -29% людей имеют эти симптомы | ||
| Нарушение минеральной плотности костной ткани | 0004348 | |
| Нарушение морфологии сердечно-сосудистой системы | 0030680 | |
| Синдактилия пальцев | 0006101 | |
| Паховая грыжа | 0000023 | |
| Гипергибкость суставов | Суставы выходят за пределы ожидаемого диапазона движений | 0005692 |
| Преаурикулярная кожная бирка | 0000384 | |
| Рецидивирующие переломы | Повышенная частота переломов Повышенные переломы Множественные переломы Множественные самопроизвольные переломы Различная степень множественных переломов [ более ] | 0002757 |
| Процент людей, у которых есть эти симптомы, недоступен через HPO | ||
| Патология почек | Аномальная почка | 0000077 |
| Аномалия ушной раковины | Уши неправильной формы Порок развития ушной раковины Деформированные уши Деформированные уши [ более ] | 0000377 |
| Агрессивное поведение | Агрессия Агрессивное поведение Агрессивность [ более ] | 0000718 |
| Нарушение прикуса переднего открытого прикуса | Отсутствие перекрытия передних верхних и нижних зубов Промежуток между верхними и нижними передними зубами при прикусывании [ более ] | 0009102 |
| Беспокойство | Чрезмерное, постоянное беспокойство и страх | 0000739 |
| Аутизм | 0000717 | |
| Двустворчатый язычок | 0000193 | |
| Катаракта | Помутнение хрусталика глаза Мутный объектив [ более ] | 0000518 |
| Явно счастливый нрав | 0100024 | |
| Крипторхизм | Неопустившиеся яички Неопустившееся яичко [ более ] | 0000028 |
| Задержка речевого и языкового развития | Нарушение речевого развития Задержка языкового развития Отложенная речь Задержка получения речи Задержка речевого развития Нарушение речи и языкового развития Нарушение речевого развития Языковая задержка Язык отложен Дефицит языкового развития Позднее развитие речи Плохое языковое развитие Задержка речи и языка Речевые и языковые трудности Задержка речи [ более ] | 0000750 |
| Диастаз прямых мышц живота | Разрыв между большими левой и правой мышцами живота | 0001540 |
| Затруднения при ходьбе | Трудности при ходьбе | 0002355 |
| Углы рта опущенные | Опущенные уголки рта Опущенный рот [ более ] | 0002714 |
| Эхолалия | Повторение речи другого человека | 0010529 |
| Асимметрия лица | Асимметрия лица Кривое лицо Несимметричное лицо [ более ] | 0000324 |
| Гримаса | 0000273 | |
| Проблемы с кормлением в младенчестве | 0008872 | |
| Функциональное нарушение дыхания | 0002795 | |
| Гастроэзофагеальный рефлюкс | Кислотный рефлюкс Кислотная рефлюксная болезнь Изжога [ более ] | 0002020 |
| Задержка роста | Задержка роста Дефицит роста Нарушение роста Задержка роста Плохой рост Замедленный рост [ более ] | 0001510 |
| Нарушение слуха | Глухота Дефект слуха [ более ] | 0000365 |
| Высокий осевой трирадиус | 0001042 | |
| Гиперактивность | Более активен, чем обычно | 0000752 |
| Гиперакузия | 0010780 | |
| Гипертония | 0001276 | |
| Гипоспадия | 0000047 | |
| Умственная отсталость | Умственная отсталость Умственная отсталость Умственная отсталость неспецифическая Умственная отсталость [ более ] | 0001249 |
| Длинная грань | Удлинение лица Увеличенная высота лица Увеличенная длина лица Вертикальное удлинение лица Вертикальное увеличение лица Вертикальное разрастание лица [ более ] | 0000276 |
| Низко посаженные уши | Низко посаженные уши Низко посаженные уши [ более ] | 0000369 |
| Приводная мышца плюсны | Передняя половина стопы поворачивается внутрь | 0001840 |
| Близорукость | Близкий вид Близорукий Близорукость Близорукость [ более ] | 0000545 |
| Узкая грань | Уменьшение ширины лица Уменьшение ширины лица [ более ] | 0000275 |
| Гипотония новорожденных | Низкий мышечный тонус в неонатальном периоде | 0001319 |
| Вызывающее оппозиционное расстройство | 0010865 | |
| Атрофия зрительного нерва | 0000648 | |
| Чрезмерное дружелюбие | 0100025 | |
| Pes planus | Плоскостопие Плоскостопие [ более ] | 0001763 |
| Преждевременное поседение волос | Раннее поседение Преждевременное поседение Преждевременное поседение Преждевременное поседение волос [ более ] | 0002216 |
| Выступающие надглазничные гребни | Выдающаяся бровь | 0000336 |
| Рецидивирующие инфекции в младенчестве и раннем детстве | 0005437 | |
| членовредительство | Умышленное членовредительство Членовредительство [ более ] | 0000742 |
| Короткий интервал внимания | Плохая концентрация внимания Проблема с вниманием [ более ] | 0000736 |
| Короткая пястная кость | Укороченная длинная кость руки | 0010049 |
| Короткая плюсневая кость | Короткая длинная кость стопы | 0010743 |
| Короткий желобок | 0000322 | |
| Одиночная поперечная ладонная складка | 0000954 | |
| Маленький для гестационного возраста | Масса тела при рождении менее 10-го процентиля Низкий вес при рождении [ более ] | 0001518 |
| спорадически | Нет предыдущего семейного анамнеза | 0003745 |
| Стеноз наружного слухового прохода | Сужение прохода от внешнего к среднему уху | 0000402 |
| Стереотипия | Повторяющиеся движения Повторяющееся или самоповреждающее поведение [ более ] | 0000733 |
| Косоглазие | Косоглазый Косоглазие Прищуренные глаза [ более ] | 0000486 |
| Синдактилия | Перепончатые пальцы рук или ног | 0001159 |
| Толстая киноварь нижней губы | Увеличение объема нижней губы Пухлая нижняя губа Выступающая нижняя губа [ более ] | 0000179 |
Круг дружбы — Блог для людей с особыми потребностями
Вы слышали о синдроме Дауна и синдроме ломкой Х-хромосомы, но как насчет синдрома Якобсена? Или 22q11.2 делеционный синдром?
В этом посте мы обращаем внимание на некоторые менее известные хромосомные нарушения.
Если у вашего ребенка есть одно из этих условий, поделитесь с нами своим опытом в комментариях ниже.
1. Синдром Вольфа-Хиршхорна
Описание: Синдром Вольфа-Хиршхорна вызывается делецией дистального короткого плеча хромосомы 4. Основные признаки расстройства включают характерный внешний вид лица, задержку роста и развития, умственную отсталость и судороги.
Организации, способствующие повышению осведомленности: Реальная история о синдроме Вольфа-Хиршхорна
В новостях: Жизнь с синдромом Вольфа-Хиршхорна
2. Синдром Якобсена
Описание: Синдром Якобсена, также известный как расстройство с делецией 11q, возникает в результате потери генетического материала на конце длинного плеча хромосомы 11. Признаки и симптомы этого состояния различаются, но у большинства людей наблюдается задержка развития двигательных навыков. речь, когнитивные нарушения, трудности с обучением и некоторые поведенческие проблемы.
Организации, способствующие повышению осведомленности: 11q Research & Resource Group
В новостях: Тысячи собраны для лечения синдрома Якобсена
3. Синдром Ангельмана
Описание: Синдром Ангельмана (AS) представляет собой пример геномного импринтинга, при котором делеция или инактивация генов на наследуемой по материнской линии хромосоме 15 вызывает импринтинг и замалчивание отцовской копии, которая может иметь нормальную последовательность. АС характеризуется задержкой умственного развития и развития, нарушениями сна, припадками и резкими движениями, но также частым смехом или улыбкой и обычно имеет счастливое поведение.
Организации, способствующие повышению осведомленности: F.A.S.T & Angelman Foundation, Inc.
В новостях: Подробно: синдром Ангельмана
4. Синдром Тернера
Описание: Синдром Тернера (TS) возникает, когда одна из двух X-хромосом у женщин отсутствует или неполна. Наиболее частыми симптомами являются низкий рост и дисгенезия гонад, которые могут вызвать неполное половое развитие, нарушение функции яичников и бесплодие.На данный момент причина TS неизвестна.
Организации, способствующие повышению осведомленности: Общество по синдрому Тернера США
В новостях: Семья распространяет информацию о синдроме Тернера
5. Синдром делеции 22q11.2
Описание: Синдром делеции 22q11.2 вызывается делецией небольшого участка 22 хромосомы около середины хромосомы. Поскольку признаки и симптомы синдрома делеции 22q11.2 разнообразны, различные группы симптомов когда-то были описаны как полностью отдельные состояния, названные синдромом ДиДжорджи, велокардиофациальным синдромом и синдромом конотрункальной аномалии лица.
Организации, способствующие повышению осведомленности: The International 22q11.2 Deletion Syndrome Foundation, Inc.
В новостях: Конференция по синдрому делеции 22q11.2, на которой собираются исследователи, преподаватели и семьи
6. Синдром тройной Икс
Описание: Синдром тройной Х-хромосомы характеризуется наличием дополнительной Х-хромосомы в каждой женской клетке. Это не вызывает каких-либо необычных физических особенностей, но связано с повышенным риском нарушения обучаемости и задержкой развития речи и языковых навыков.
Организации, способствующие повышению осведомленности: Синдром Triple X
В новостях: Что такое синдром Triple X? Что вызывает синдром тройного Икс?
7. Синдром Вильямса
Описание: Синдром Вильямса вызывается делецией генетического материала из частей длинного плеча хромосомы 7, области, которая состоит из более чем 25 генов. Исследователи идентифицировали несколько конкретных генов, связанных с синдромом Вильямса, но связь между большинством генов в удаленной области и симптомами синдрома Вильямса до сих пор неизвестна.
Организации, способствующие повышению осведомленности: Ассоциация синдрома Вильямса
В новостях: Синдром Вильямса показывает связь между генами и поведением: ключ к аутизму?
8. Синдром Кри дю Чат
Описание: Синдром Кри-дю-Шат возникает из-за отсутствия части хромосомы 5. Симптомы включают пронзительный крик, похожий на кошачий, наклон глаз вниз, частичное переплетение перепонок или слияние пальцев рук или ног, а также медленный или неполный развитие моторики.
Организации, способствующие повышению осведомленности: Общество Five P минус
В новостях: Синдром «кошачьего крика», часто не диагностируемый
9. Трисомия 13 / синдром Патау
Описание: Трисомия 13, также называемая синдромом Патау, представляет собой заболевание, при котором у человека есть три копии генетического материала из хромосомы 13, а не две. Это может произойти в трех формах: трисомия 13, которая имеет третью хромосому 13 во всех клетках; Мозаицизм трисомии 13, который имеет третью хромосому 13 в некоторых клетках; и частичная трисомия, при которой в клетках присутствует часть дополнительной хромосомы 13.
Организации, способствующие повышению осведомленности: Жизнь с трисомией 13
В новостях: Sequenom проверяет MaterniT21 для обнаружения трисомии 18 и 13
10. Трисомия 18 / синдром Эдвардса
Описание: Трисомия 18, или синдром Эдвардса, возникает, когда человек имеет третью копию материала хромосомы 18 вместо обычных двух копий. Некоторые симптомы включают сжатые руки, ступни с закругленным дном, умственную отсталость, недоразвитые ногти и грудную клетку необычной формы.
Организации, способствующие повышению осведомленности: Фонд Трисомии 18
В новостях: История малышки Джалиссы: борьба с трудностями, жизнь с синдромом Эдварда
11. Синдром кошачьего глаза
Описание: У людей с синдромом кошачьего глаза короткое плечо (известное как 22p) и небольшая область длинного плеча (22q) хромосомы 22 присутствуют три или четыре раза, а не дважды. Характерные признаки расстройства включают умеренную задержку роста до рождения, умеренную умственную отсталость и пороки развития навыков и лицевой области, сердца, почек и / или анальной области.
Организации, способствующие повышению осведомленности: Поддержка хромосомных заболеваний 22
В новостях: Блог: «Наше путешествие с синдромом кошачьего глаза»
12. Трисомия 16
Описание: Полная трисомия 16 возникает, когда у человека есть три копии хромосомы 16 вместо обычных двух, и является наиболее частой хромосомной причиной выкидыша в первом триместре беременности. Мозаичная трисомия 16 — это редкое заболевание, при котором дополнительная хромосома 16 присутствует в некоторых клетках, но не во всех.Некоторые общие симптомы включают задержку внутриутробного развития (ЗВУР) и врожденные пороки сердца.
Организации, способствующие повышению осведомленности: Расстройства хромосомы 16
В новостях: Трисомия 16: основная причина выкидыша
13. Болезнь Шарко-Мари-Тута
Описание: Болезнь Шарко-Мари-Тута (ШМТ) является наиболее распространенным наследственным неврологическим заболеванием, вызываемым генетическими мутациями. CMT1A является результатом дупликации гена на хромосоме 17, который несет инструкции по производству периферического миелина протенина-22.
Организации, способствующие повышению осведомленности: Ассоциация Шарко-Мари-Тута
В новостях: Причина заболевания определяется геномом
Понравился пост? Закрепите его для дальнейшего использования на Pinterest здесь:
NZ Herald — Последние новости, последние новости бизнеса, спорта и развлечений
Похоже на тупик.
Сделайте резервную копию или перейдите на нашу домашнюю страницу….
Певец в маске: опоссум в свете фар и сборка Мстителей
3 минуты на чтение
Эпизод 6 здесь с настолько радикальными изменениями, что мы просто не можем их игнорировать.
«Он был моим младенцем»: мать убитого 6-летнего человека раскрывает последние моменты жизни со своим сыном
4 минуты на чтение
Эйден Леос был застрелен в результате перестрелки на дороге в Калифорнии.
Premium
Оговорка «торговец наркотиками» после того, как полиция заморозила активы из-за работы со смертельным исходом
7 минут на чтение
Босс рассказывает о влиянии на бизнес дела о «доходах от преступлений».
Мужчина серьезно ранен в результате ножевого ранения в автобусе в Западном Окленде.
Краткое прочтение.
Шокированный водитель автобуса вызвал полицию после ранения пассажира.
От «очень плохого» воспитания до генерал-губернатора, история госпожи Синди Киро
4 минуты на чтение
«Я надеюсь, молодые девушки маори … и все девушки, возьмите немного вдохновение », — говорит дама Синди Киро.
Premium
Начальник вернулся, но рабочие остались дома
7 минут на чтение
Руководители, вернувшиеся в офис, изо всех сил пытаются убедить сотрудников присоединиться к ним.
Premium
Школа оплакивает первокурсника, «не имеющего отношения к жизни»
5 минут на чтение
«Один учитель, или человек, или образец для подражания, которого дети никогда не станут, когда-нибудь забыть.»
Premium
Закрытие рынка: Брокер сдерживает импульс Раймана; рынок акций падает
4 минуты на чтение
У рынка акций была плохая дневная сессия, падение 0,09% за день.
Безмерная «грусть» королевы из-за того, что она никогда не видела Арчи
2 минуты на чтение
Разлучение с внуком вызвало у Монарха крайнюю «печаль», — говорит эксперт
.