Нарушения минерального обмена в почках (дисметаболические нефропатии)
Дисметаболические нефропатии (наршения минерального обмена почек — НМО) представляют собой группу заболеваний, характеризующихся поражением почек вследствие нарушения обмена веществ и приводящим к развитию мочекаменной болезни (МКБ), воспалению почек (пиелонефрит), которые могут осложниться хронической почечной недостаточностью (ХПН).
В зависимости от причины развития выделяют первичные и вторичные НМО.
Первичные нарушения представляют собой наследственно обусловленные формы заболеваний, характеризующихся прогрессирующим течением, ранним развитием мочекаменной болезни (МКБ) и хронической почечной недостаточности (ХПН). Первичные дисметаболические нефропатии встречаются редко и начало клинических проявлений развивается уже в детском возрасте.
Вторичные дисметаболические нефропатии могут быть связаны с повышенным поступлением определенных веществ в организм, нарушением их метаболизма в связи с поражением других органов и систем (например, желудочно-кишечного тракта), применением ряда лекарственных средств и др.
Подавляющее большинство (от 70 до 90%) дисметаболических нефропатий связаны с нарушением обмена кальция, при этом около 85–90% из них обусловлены избытком солей щавелевой кислоты в виде оксалата кальция — оксалатов, 3–10% — перегрузкой фосфатами (фосфаты кальция), существуют смешанный вариант нарушений – оксалатно/фосфатно-уратные.
Оскалатно-кальциевая нефропатия
Оксалатно-кальциевая нефропатия наиболее часто встречается в детском возрасте. Ее возникновение может быть связано с нарушением обмена кальция или оксалатов (солей щавелевой кислоты).
Причины образования оксалатов:
- повышенное поступление оксалатов с пищей
- заболевания кишечника – воспалительные заболевания кишечника (болезнь Крона, язвенный колит), кишечные анастомозы при проведении оперативных вмешательств на кишечнике
- повышенная выработка оксалатов самим организмом
Оксалатная нефропатия представляет собой многофакторный патологический процесс. Наследственная предрасположенность к развитию оксалатной нефропатии встречается в 70–75%. Помимо генетических, большую роль играют такие внешние факторы, как: питание, стресс, экологические проблемы и др.
Наследственная предрасположенность к развитию оксалатной нефропатии встречается в 70–75%. Помимо генетических, большую роль играют такие внешние факторы, как: питание, стресс, экологические проблемы и др.
Первые проявления болезни могут развиться в любом возрасте, даже в период новорожденности. Чаще всего они выявляются в 5–7 лет в виде обнаружения кристаллов оксалатов, небольшим содержанием белка, лейкоцитов и эритроцитов в общем анализе мочи. Характерно повышение удельной плотности мочи. Заболевание обостряется в период полового созревания в возрасте 10–14 лет, что, по-видимому, связано с гормональной перестройкой.
Прогрессирование оксалатной нефропатии может привести к формированию мочекаменной болезни, развитию воспаления почек — пиелонефриту при наслоении бактериальной инфекции.
Фосфатная нефропатия
Фосфатная нефропатия встречается при заболеваниях, сопровождающихся нарушением фосфорного и кальциевого обмена. Основная причина фосфатурии – хроническая инфекция мочевой системы. Часто фосфатно-кальциевая нефропатия сопровождает оксалатно-кальциевую, но при этом выражена в меньшей степени.
Уратная нефропатия (нарушения обмена мочевой кислоты)
Эта группа обменных нарушений наиболее часто встречается у взрослых. Первичные уратные нефропатииобусловлены наследственными нарушениями обмена мочевой кислоты. Вторичные возникают как осложнениядругих заболеваний (болезней крови и др.), являются следствием применения некоторых препаратов (тиазидовых диуретиков, цитостатиков, салицилатов, циклоспорина А и др.) или нарушения функции канальцев почек и физико-химических свойств мочи (при воспалении почек, например). Кристаллы уратов откладываются в ткани почек – это приводит к развитию воспаления и снижению почечных функций.
Первые признаки заболевания могут выявляться в раннем возрасте, хотя в большинстве случаев наблюдается длительное скрытое течение процесса.
Цистиновая нефропатия
Цистин является продуктом обмена аминокислоты метионина. Можно выделить две основные причины повышения концентрации цистина в моче:
Можно выделить две основные причины повышения концентрации цистина в моче:
- избыточное накопление цистина в клетках почки
- нарушение обратного всасывания цистина в почечных канальцах.
Накопление цистина в клетках происходит в результате генетического дефекта фермента цистинредуктазы. Это нарушение обмена носит системный характер и называется цистинозом. Внутриклеточное и внеклеточное накопление кристаллов цистина выявляется не только в канальцах и интерстиции почки, но и в печени, селезенке, лимфоузлах, костном мозге, клетках периферической крови, нервной и мышечной ткани, других органах. Нарушение обратного всасывания цистина в канальцах почек наблюдается вследствие генетически обусловленного дефекта транспорта через клеточную стенку для аминокислот – цистина, аргинина, лизина и орнитина.
По мере прогрессирования заболевания определяются признаки мочекаменной болезни, а при присоединении инфекции – воспаление почек.
Симптомы НМО
НМО почек, как правило, клинически течет бессимптомно до формирования мочекаменной болезни или пиелонефрита, но в ряде случаев могут проявляться следующими симптомами:
- дискомфорт при мочеиспускании
- учащенное мочеиспускание
- ноющая боль или дискомфорт, локализующиеся преимущественно в поясничной области или животе
- приступообразная («почечная колика») или постоянная боль, отдающая в. подвздошную или паховую область, на внутреннюю поверхность бедра, в половые органы
- боль над лоном может развиваться при отложении солей или наличии камней в мочевом пузыре
Диагностика НМО
Необходимое комплексное обследование включает лабораторные и инструментальные методы.
Лабораторная диагностика
- Общий анализ мочи, в котором выявляются кристаллы солей той или иной кислоты. Однако данное исследование не позволяет с полной уверенностью утверждать о присутствии дисметаболической нефропатии. Выявление кристаллов солей только в общих анализах мочи не является основанием для постановки диагноза дисметаболической нефропатии.
 Следует иметь в виду, что выделение кристаллов с мочой часто бывает преходящим и оказывается не связанным с нарушением обмена веществ. Поэтому для уточнения диагноза прибегают ко второму этапу исследования – проведению биохимического исследования мочи.
Следует иметь в виду, что выделение кристаллов с мочой часто бывает преходящим и оказывается не связанным с нарушением обмена веществ. Поэтому для уточнения диагноза прибегают ко второму этапу исследования – проведению биохимического исследования мочи. - Биохимический анализ мочи позволяет оценить концентрации тех или иных солей в порции мочи. Метод является более точным и чувствительным для определения количественного уровня оксалатов, фосфатов, уратов и других кристаллов солей.
- АКОСМ — определение антикристаллобразующей способности мочи. Метод достаточно сложный, проводится не в каждом лечебном заведении.
- Ряд тестов на перекиси в моче и кальцифилаксию. Данная методика позволяет выявить нарушения кальциевого обмена и оценить степень активности перекисного окисления мембран клеток почечной ткани, что является важным звеном в процессах развития дисметаболических нефропатий.
 Следует иметь в виду, что выделение кристаллов с мочой часто бывает преходящим и оказывается не связанным с нарушением обмена веществ. Поэтому для уточнения диагноза прибегают ко второму этапу исследования – проведению биохимического исследования мочи.
Следует иметь в виду, что выделение кристаллов с мочой часто бывает преходящим и оказывается не связанным с нарушением обмена веществ. Поэтому для уточнения диагноза прибегают ко второму этапу исследования – проведению биохимического исследования мочи.Инструментальная диагностика
УЗИ органов брюшной полости. Изменения, выявляемые при УЗИ почек, как правило, мало специфичны. Возможно выявление в почке микролитов или «песка» (включений). УЗИ почек, как правило, является неспецифичным методом диагностики, однако в ряде случаев позволяет отследить формирование мелких камней и, таким образом, зафиксировать время возникновения мочекаменной болезни.
Лечение
Рекомендации по питанию являются основой терапии как в детском, так и во взрослом состоянии.
| Вид нефропатии | Запрещенные продукты |
| Оксалатная нефропатия | Мясные блюда, щавель, шпинат, клюква, свекла, морковь, какао, шоколад |
| Уратная нефропатия | Печень, почки, мясные бульоны, горох, фасоль, орехи, какао, алкогольные напитки |
| Фосфатная нефропатия | Сыр, печень, икра, курица, бобовые, шоколад |
| Цистиновая нефропатия | Творог, рыба, яйца, мясо |
Лечение оксалатной нефропатии
Диета
- при лечении больных с оксалатной нефропатией назначается картофельно-капустная диета, при которой снижается поступление оксалатов с пищей и нагрузка на почки
- необходимо исключить холодец, крепкие мясные бульоны, щавель, шпинат, клюкву, свеклу, морковь, какао, шоколад
- рекомендуется ввести в рацион курагу, чернослив, груши
- из минеральных вод используются такие, как славяновская и смирновская, по 3–5 мл/кг/сут. в 3 приема курсом 1 месяц 2–3 раза в год
 в 3 приема курсом 1 месяц 2–3 раза в год
в 3 приема курсом 1 месяц 2–3 раза в годЛекарственная терапия включает мембранотропные препараты и антиоксиданты. Лечение должно быть длительным. Применяются витамины группы В, А, Е. Назначаются специальные препараты при кристаллурии. Помимо этого, назначается окись магния, особенно при повышенном содержании оксалатов.
Лечение уратной нефропатии
Диета
- при лечении уратной нефропатии диета предусматривает исключение богатых пуриновыми основаниями продуктов (печени, почек, мясных бульонов, гороха, фасоли, орехов, какао и др.)
- преимущество должно отдаваться продуктам молочного и растительного происхождения
- важным условием успешной терапии является достаточное употребление жидкости – от 1 до 2 л в сутки. Предпочтение следует отдавать слабощелочным и слабоминерализованным водам, отварам трав (хвощ полевой, укроп, лист березы, брусничный лист, клевер, спорыш и др.), отвару овса
Для поддержания оптимальной кислотности мочи можно использовать цитратные смеси. При уратной нефропатии важно уменьшить концентрацию мочевой кислоты. Для этого используются средства, снижающие синтез мочевой кислоты.
Лечение фосфатной нефропатии
Назначается диета с ограничением продуктов, богатых фосфором (сыр, печень, икра, курица, бобовые, шоколад и др.).
Лечение при фосфатной нефропатии должно быть направлено на подкисление мочи (минеральные воды – нарзан, арзни, дзау-суар и др.; препараты – цистенал, аскорбиновая кислота, метионин).
При любой степени тяжести заболевания необходимо незамедлительно обратиться к врачу нефрологу или урологу за помощью, так как длительно текущие, в целом обратимые, нарушения обмена при отсутствии лечения могут привести к развитию мочекаменной болезни с последующим оперативным вмешательством и хронической почечной недостаточности. Самолечение не допустимо!
Все виды лекарственной терапии должен назначать и обязательно контролировать врач нефролог или уролог, так как:
- эти лекарства имеют серьезные побочные эффекты на другие органы и системы
- у части пациентов отмечается изначальная невосприимчивость или постепенно развивается устойчивость к препаратам
На первом этапе лечения составляется план лечения. Лечение любой дисметаболической нефропатии можно свести к четырем основным принципам:
Лечение любой дисметаболической нефропатии можно свести к четырем основным принципам:
- нормализация образа жизни
- правильный питьевой режим
- диета
- специфические методы терапии
Прием большого количества жидкости является универсальным способом лечения любой дисметаболической нефропатии, так как способствует уменьшению концентрации растворимых веществ в моче.
Одной из целей лечения является увеличение ночного объема мочеиспускания, что достигается приемом жидкости перед сном. Предпочтение следует отдавать простой или минеральной воде.
Диета позволяет в значительной степени снизить солевую нагрузку на почки.
Специфическая терапия должна быть направлена на предупреждение конкретного кристаллообразования, выведение солей, нормализацию обменных процессов.
На втором этапе терапии производится оценка эффективности диеты, проводятся контрольные УЗ-исследования и анализы.
Третий этап лечения осуществляется после достижения стойкой ремиссии. Он представляет собой схему постепенного снижения доз назначенных препаратов до поддерживающих или полной их отмены с сохранением диетических рекомендаций.
Даже после достижения долгожданной ремиссии пациенту рекомендуется быть внимательным к себе и регулярно наблюдаться у врача нефролога или уролога, так как высок риск рецидива заболеваний.
Практически всем пациентам необходимо принимать рекомендованные врачом средства противорецидивной терапии или придерживаться ранее разработанной диеты для предотвращения формирования или прогрессирования МКБ, воспаления почек.
Прогноз
Прогноз при дисметаболической нефропатии в целом благоприятен. В большинстве случаев при соответствующем режиме, диете и лекарственной терапии удается добиться стойкой нормализации соответствующих показателей в моче. В отсутствие лечения или при его неэффективности наиболее естественным исходом дисметаболической нефропатии является мочекаменная болезнь и воспаление почек.
Самым частым осложнением дисметаболической нефропатии является развитие инфекции мочевой системы, в первую очередь пиелонефрита.
Если вы обнаружили у себя какие-либо из перечисленных выше симптомов (нарушение мочеотделения, изменения свойств мочи, боли), необходимо в ближайшее время обратиться к врачу за помощью.
Помните, что очень важно начать лечение на ранних стадиях болезни, так как НМО в почках является преимущественно обратимым состоянием, а в случае отсутвтия лечения итогом является развитие мочекаменной болезни, пиелонефрита.
Рекомендации
Для профилактики развития заболевания, а так же рецидива, необходимо придерживаться правильного, сбалансированного и регулярного питания – избегать острой пищи, маринадов и пр. В период обострения пациентам рекомендуется щадящая диета, соответствующая требованиям биохимического вида нефропатии (оксалатная, уратная и пр.).
С целью профилактики рецидивов всем пациентам рекомендуется один раз в квартал консультация врача нефролога или уролога для необходимой коррекции медикаментозной терапии и пищевых рекомендаций.
Пациенты, длительно страдающие НМО в почках, относятся к группе повышенного риска по МКБ. Поэтому в период ремиссии им необходимо ежегодно проходить по назначению врача контрольные обследования (общий анализ мочи, биохимия мочи, УЗИ почек, мочеточников, мочевого пузыря) мочевыводящей системы.
Часто задаваемые вопросы
Излечимы ли НМО?
НМО часто обусловлено наследственными нарушениями обмена, что требует постоянного соблюдения как минимум диетических рекомендаций.
Из-за чего возникает заболевание?
НМО может быть связано с наследственной предрасположенностью, а также с заболеваниями внутренних органов (желудочно-кишечные проблемы, заболевания крови и пр.), применением определенных групп лекарственных препаратов (мочегонные средства, цитостатики и пр.).
Является ли заболевание противопоказанием к беременности?
Само НМО в почках требует наблюдения весь период беременности с соблюдением диетических рекомендаций.
При развитии МКБ, пиелонефрита и их осложнений в виде ХПН, возможность беременности и ее сохранения зависит от обострения процесса и стадии осложнений и решается в каждом конкретном случае.
Может ли заболевание почек проявляться снижением потенции?
Непосредственно НМО конечно не влияет на потенцию, но в случае развития осложнений МКБ, воспаления почек или развития ХПН снижение потенции может появиться как реакция на хроническое заболевание.
Увеличивается ли риск заболеть, если близкий родственник страдает данным заболеванием?
Да, существует группа первичных обменных нефропатий (НМО в почках), имеющая наследственную предрасположенность.
код по мкб 10, причины и лечение
Информация носит справочный характер. Не занимайтесь самодиагностикой и самолечением. Обращайтесь ко врачу.
Почки являются фильтром жидкостей в организме человека.
Вследствие многих факторов возникают болезни этих органов.
Патология поражает эпителий почек, и из-за этого в органе скапливаются соли мочевой и щавелевой кислоты.
Эти процессы приводят к заболеванию дисметаболической нефропатии.
Содержание статьи
Основная информация и код по мкб-10
Патология заключается в комплексе болезней. Скопление определённых веществ в эпителии почек наблюдаются вследствие нарушения метаболического процесса.
Причины появления отложения солей и других веществ. Это считается важным фактором, что предрасполагает к развитию болезни по 2 типам:
- Наследственные заболевания с прогрессивным течением. Патологии развиваются рано и к ним относят мочекаменную болезнь и почечную недостаточность в хронической форме.
- Возникают из-за увеличения поступления веществ, и нарушается метаболизм вследствие поражения других органов.
У дисметаболической нефропатиии код по Международному классификатор болезней десятого пересмотра, имеет номер 16.3
Статистика заболевания
Патология проявляется из-за следующих факторов:
- обменных процессов кальция от 70% до 90% случаев у детей и взрослых;
- избытка солей щавелевой и мочевой кислоты от 85% до 90% случаев оксалурии;
- избытка фосфата кальция наблюдаются от 3% до 10% больных.

Проявление недуга у новорожденных зависит от наследственных отклонений.
Общие причины возникновения
Появление патологии происходит вследствие снижения метаболизма организма. Помимо этого, причиной заболевания выступают воспалительные процессы мочеполовой системы.
Нарушение метаболизма эндокринной патологии, что приводит к нефропатии. С фактором развития, иногда связаны другие заболевания органов. К этому относят нарушения в работе желудочно-кишечного тракта.
Если ранее происходило лечение лучевой терапией, то болезнь возникает как следствие онкологического заболевания.
Когда человек питается неправильно, и не имеет определённого рациона питания, то в организме накапливаются оксалаты и ураты. Их повышенная концентрация вызывает нарушение в работе почек.
Этиология у детей
Заболевание у детей может проявляться врождённой патологией. Это происходит из-за позднего токсикоза и гипоксии плода.
Помимо этого возникает заболевание, которое проявляется вследствие наследственного нарушения обменных процессов организма.
В зависимости от вида поражения почек, болезнь появляется по следующим причинам:
- нарушается метаболизм кальция и оксалатов;
- в организме уменьшается количество витаминов групп А, В6 и Е;
- в дефиците находится магний и калий;
- возрастает количество концентрации витамина D.
Эти факторы вызывают оксалатную дисметаболическую нефропатию. Происхождение приведённых причин, связывают с следующими ппатологиями:
- болезни Крона;
- энтерита;
- колита;
- хронического панкреатита;
- дискинезии желчевыводящих путей;
- сахарного диабета;
- пиелонефрита.
Появление патологии обусловлено лечением препаратами, в состав которых входят циостатики, тиазидовые диуретики и салицилаты.
В иных случаях заболевание нефропатии происходит по следующим причинам:
- хронической мочевой инфекции;
- гиперпаратиреоза;
- заболеваний центральной нервной системы.

Возникновение болезни у детей связано с пиелонефритом. Происходит избыточное накопление веществ с нехваткой других элементов в организме.
С чем связано возникновение у взрослых?
Взрослые заболевают патологией по таким причинам:
- нарушение обмена веществ вследствие несбалансированного питания;
- проявление влияния из-за ионного излучения;
- употребление воды и жидкости с повышенным содержанием кальция;
- нарушение в работе и патологии щитовидной железы;
- заболевания внутренних органов, которые протекают в хронической форме.
Если взрослый человек постоянно употребляет кофе, то такая ситуация предоставляет возможность развитию дисметаболической нефропатии. При чрезмерном употреблении слабоалкогольных напитков возникновении патологии имеет высокую вероятность.
Это касается постоянное принятие в пищу мяса. Кишечник и желудок не успевают выводить избыток минеральных отложений. Тогда вещества оседают в тканях и других органах.
Классификация и виды
Существует множество разновидностей заболевания. К ним можно отнести:
- Оксалатно-кальциевая нефропатия – способна проявляться в детском возрасте. Возникновение обусловлено нарушением метаболизма кальция и щавелевой кислоты.
- Оксалатнуая разновидность – проявляется в любом возрасте, иногда возникает сразу же после рождения. В большинстве случаев наблюдается у детей от 7 лет. Образующиеся кристаллы обнаруживают в урине с помощью анализа. Отличительной чертой считается вязкая моча. Обострение возникает из-за полового созревания от 13 лет.
- Фосфатная – проявляется вследствие нарушения метаболизма фосфатов и кальция.
- Уратная – в течение суток организм вырабатывает 10 мл мочевой кислоты. Часть от вещества попадает в кишечник и происходит процесс нейтрализации микроорганизмами. Оставшаяся часть урины очищается в почках и возвращается. Кристаллы оседают в органе и образуются камни.
- Цистинозный вид – происходит увеличение содержания цистина в урине. Скопление вещества возникает из-за наследственного отклонения. Оседание кристаллов наблюдается не только в почках, но и в печени, селезёнке, в тканях нервов и мышц. Со временем отмечается возникновение камней и проявление воспалительных процессов в органах.
 Кристаллы оседают в органе и образуются камни.
Кристаллы оседают в органе и образуются камни.Симптоматика в представленных классификациях патологии отличается у детей и взрослых.
Симптомы проявления
Дисметаболическая нефропатия на ранних этапах развития не проявляется в видимых симптомах. В дальнейшем, признаки у детей и взрослых отличаются.
Признаки у детей
Симптомы у детей проявляются в следующем:
- консистенция мочи;
- повышенное или пониженное артериальное давление;
- отёки век;
- повышенная потливость;
- боли в животе;
- постоянная жажда или сухость во рту;
- нет аппетита;
- головные боли;
- кожа ребёнка сухая.
Особенность патологии считается в выделении густой мочи. Можно наблюдать образование кристаллов. Многие дети страдают повышенным давлением.
Для обнаружения заболевания могут способствовать препараты, которые не способны вылечить этот симптом.
Утром у ребёнка может наблюдаться отёчность век. Затем этот признак переходит на нижние конечности, пальцы, руки и ноги.
Некоторые дети страдают заболеванием с рождения. Симптомы не проявляются, и большую часть жизни нефропатия протекает латентно. В семьях обнаруживаются больные артропатией, падагрой, спондилёзом, мочекаменной болезнью и сахарным диабетом.
У взрослых
Взрослые страдают нарушением метаболизма, и проявлением болезни становится накопление солевых отложений в почках. Кристаллы образуются в камни и песок, тем самым закупоривают мочеточник.
Когда происходит процесс выделения, то они начинают царапать ткани. Это вызывает болевые ощущения. Основными симптомами дисметаболической нефропатии у взрослых считаются:
- дизурия;
- боль в голове и суставах;
- частое пониженное давление;
- болевой синдром при поколачивании поясницы;
- аппетита нет;
- шелушение кожи;
- постоянная потливость;
- недержание мочи;
- запоры.

В отличие от детей у взрослых артериальное давление понижено.
К кому обратиться за диагностикой?
Если обнаружились симптомы заболевания необходимо обратиться за диагностикой к специалисту. Для детей обследование проводит детский уролог или нефролог.
Затем его направляют на исследование антикристаллобразующие способности мочи, чтобы подтвердить диагноз дисметаболической нефропатии. Заключительной диагностикой считается ультразвуковое обследование почек.
Способы терапии
Заболевание не является опасным, но доставляет дискомфорт. Поэтому нужно следовать рекомендациям врача. Лечение дисметаболической нефропатии детей и взрослых имеет свою специфику. Решающим фактором для назначения терапии является вид патологии.
Лечение детей
Если у ребёнка обнаружена оксалурия, то назначается частое употребление жидкости. При отложении солей в почках за сутки требуется выпивать не менее 1,5 л воды. Воду дают ребёнку на ночь.
Для этого лучшим средством будет минералка. Можно давать ребёнку компот, клюквенный морс. Положительное действие оказывает травяной чай.
Лечебная диета состоит из продуктов, которые не образуют соли щавелевой кислоты. Лечащий врач назначает использование витаминов групп B6, А и Е. Выписываются лекарства, в состав которых входит калий и магний.
Если у ребёнка уретрия, то исключают употребление жидкости, которая способствует образованию мочевой кислоты.
Лекарства прописывают, которые снижают образование уратов. Специалисты обращают внимание на применение витаминных комплексов.
Способы для взрослых
Для проведения терапии патологии у взрослых используют лечебную диету. Из рациона требуется исключить употребление овощей, зелени, ягод и какао-бобов.
В зависимости от вида заболевания назначается объём суточной нормы выпиваемой жидкости. В основном рекомендуется обильное питьё. Это советуют для того, чтобы мочевой пузырь смог наполниться перед сном.
Во время диеты можно употреблять картошку и капусту. Из ягод и фруктов разрешается, есть чернослив, курагу и грушу.
Из ягод и фруктов разрешается, есть чернослив, курагу и грушу.
Как и в остальных случаях, организм необходимо подпитывать витаминами. Вследствие индивидуальных особенностей взрослым выписывают окись магния.
Возможные осложнения
Осложнения могут возникать, если патология проходит бессимптомно. Это приводит к следующим заболеваниям:
При несвоевременном лечении нефропатия предоставляет возможность для постоянного образования камней в почках.
Профилактика заболевания
Чтобы предотвратить появление патологии рекомендуют следующее:
- проводить диетотерапию;
- если организм подвержен заболеванию, то необходимо высчитывать содержание щавелевой кислоты в продуктах;
- употребление витаминов групп В6, С и Е;
- для профилактики фосфатов употребляют фрукты и ягоды.
Если возникли проблемы, то необходимо обратиться за помощью к педиатру, гастроэнтерологу или нефрологу.
Прогноз и заключение
Патология излечима и прогноз благоприятен. Если больной будет соблюдать все предписания лечащего врача, то организм начнёт восстанавливаться.
При неэффективности терапии может возникнуть мочекаменная болезнь. В большинстве случаев нефропатия переходит в пиелонефрит. Заболевание способно возникнуть с самого рождения. Однако протекать будет в латентной форме.
Если родители часто водят ребёнка на профилактическое обследование, то выявить дисметаболическую нефропатию будет несложно. Это заболевание требует своевременного лечения, чтобы не допустить осложнений.
Дисметаболическая нефропатия. Обменная нефропатия у детей и взрослых, лечение в СПБ
Дисметаболическая (обменная) нефропатия
Актуально для почек: обменная нефропатия и ее лечение
Обменной, или дисметаболической, нефропатией врачи именуют комплекс заболеваний почек, каждое из которых может быть вызвано нарушением обмена веществ. Ему подвержены как взрослые, так и малыши. Более того, именно дети и даже новорожденные попадают в группу риска с большей вероятностью, потому что во многих случаях недуг возникает на фоне неблагоприятной наследственности и проявляется в детском и подростковом возрасте.
Ему подвержены как взрослые, так и малыши. Более того, именно дети и даже новорожденные попадают в группу риска с большей вероятностью, потому что во многих случаях недуг возникает на фоне неблагоприятной наследственности и проявляется в детском и подростковом возрасте.
В процессе развития заболевания в почках значительно увеличивается концентрация солей, которые выделяются вместе с мочой. Это приводит к поражению не только почек, но и наружных половых органов.
Первичная обменная нефропатия нередко возникает как следствие почечной недостаточности, образования камней в мочевыводящей системе, а также на фоне иных хронических процессов. Вторичную, как правило, провоцируют неправильное питание, а значит неправильные обменные процессы, потребление большого числа медикаментов или прием их с нарушением инструкций. Довольно часто нефропатию вызывает нарушение концентрации и миграции в организме таких чрезвычайно полезных веществ и соединений, как кальций, оксалаты (соли щавелевой кислоты), фосфаты, цистин и мочевая кислота.
Причины нефропатии
- малая интенсивность обменной деятельности организма;
- воспаления органов мочеполовой системы различных типов;
- патологии эндокринного характера;
- хронические заболевания и патологии желудочно-кишечного тракта;
- загрязнение окружающей природной среды;
- недостаток или избыток микроэлементов в организме;
- употребление в пищу продуктов с высоким содержанием химических добавок;
- жесткая вода и прочие негативные факторы.
Нефрологи и урологи клиники «Долголетие» проводят тщательное диагностирование заболеваний почек. Пациенты направляются на все необходимые исследования, которые также можно пройти в нашем медцентре. В том числе сдают биохимический анализ мочи, который помогает выявить превышенную концентрацию определенных солей в моче, если нужно, проходят ультразвуковую диагностику почек, органов малого таза, мочевыводящих путей.
Симптомы обменной нефропатии
- повышенная утомляемость;
- частое и обильное мочеиспускание;
- боли внизу живота;
- отеки;
- воспаление наружных половых органов;
- пониженное артериальное давление и так далее.

Иногда наблюдаются такие проявления болезни, как кожные высыпания, зуд, а также повышение массы тела вследствие избыточного отложения жировой ткани.
Подобным симптомам нужно уделить пристальное внимание, так как вследствие обменной нефропатии могут развиться пиелонефрит, цистит, мочекаменная болезнь, нефрит и ряд других опасных заболеваний.
Для выявления, правильного диагностирования и адекватного лечения заболевания необходимо своевременно обратиться к профессионалам. Урологи, андрологи, нефрологи клиники «Долголетие» проводят консультативные приемы и осмотры на высоком квалифицированном уровне, а назначая лечение, всегда учитывают индивидуальные особенности конкретного случая и общее состояние здоровья пациента. Вовремя оказанная помощь позволит Вам быстро распрощаться с недугом и предотвратить его последствия. Чтобы записаться на прием, звоните по номеру колл-центра: 671-01-70.
Диабетическая нефропатия мкб код 10
ПОДРОБНЕЕ
была проблема- ДИАБЕТИЧЕСКАЯ НЕФРОПАТИЯ МКБ КОД 10 — Вылечила сама, смотри, что сделать-
в том числе, возникшее на фоне длительной гликемии. Заболевание выступает в качестве осложнения диабета 1 или 2 типа, вызвавший диабет, нарушаются фильтрующие Дисметаболическая нефропатия код по МКБ-10 имеет N16.3 (тубулоинтерстициальное поражение почек Для диабетической нефропатии по МКБ 10 используется 2 шифра. Поэтому диабетическая нефропатия код по МКБ 10 может иметь как Е10142 (сахарный диабет с поражением почек) МКБ-10 внедрена в практику здравоохранения на всей территории РФ в 1999 году приказом Минздрава России от 27.05.97г. 170. Для диабетической нефропатии по МКБ 10 используется 2 шифра. Поэтому диабетическая нефропатия код по МКБ 10 может иметь как Е.10-14.2 (сахарный диабет с поражением Код патологии по МКБ-10 Е10 14.2. Это позднее осложнение сахарного диабета любого типа, а также клубочков, которое встречается у 20 Диабетическая нефропатия смертельно опасное осложнение диабета. Чтобы не допустить развитие патологии, код по мкб 10. Что провоцирует образование белка в урине?
Заболевание выступает в качестве осложнения диабета 1 или 2 типа, вызвавший диабет, нарушаются фильтрующие Дисметаболическая нефропатия код по МКБ-10 имеет N16.3 (тубулоинтерстициальное поражение почек Для диабетической нефропатии по МКБ 10 используется 2 шифра. Поэтому диабетическая нефропатия код по МКБ 10 может иметь как Е10142 (сахарный диабет с поражением почек) МКБ-10 внедрена в практику здравоохранения на всей территории РФ в 1999 году приказом Минздрава России от 27.05.97г. 170. Для диабетической нефропатии по МКБ 10 используется 2 шифра. Поэтому диабетическая нефропатия код по МКБ 10 может иметь как Е.10-14.2 (сахарный диабет с поражением Код патологии по МКБ-10 Е10 14.2. Это позднее осложнение сахарного диабета любого типа, а также клубочков, которое встречается у 20 Диабетическая нефропатия смертельно опасное осложнение диабета. Чтобы не допустить развитие патологии, код по мкб 10. Что провоцирует образование белка в урине?
Впервые была оглашена мкб 10 в 1999 году, реже узелковый (хотя именно узелковый Диетотерапия при развитии диабетической нефропатии править править код . См. также: Диетотерапия сахарного диабета. На фоне слабой компенсации сахарного диабета у 10 20 пациентов развивается опасное осложнение диабетическая нефропатия (код по МКБ 10 N08.3). На фоне повреждения малых и крупных сосудов страдают многие органы, при которой уменьшается скорость фильтрации в клубочках почек (СКФ). СКФ лежит в основе деления Сахарный диабет способен вызвать различного рода осложнения и одно из них — диабетическая нефропатия. Комплекс различных поражений фильтрующих почечных элементов (канальцев и клубочков) и сосудов. Для диабетической нефропатии по МКБ 10 используется 2 шифра. Поэтому диабетическая нефропатия код по МКБ 10 может иметь как Е.10-14.2 (сахарный диабет с поражением почек), инфекцию мочевыводящих путей и паппилярный некроз. Заболевание позиционируется на первом месте по частоте развития почечной недостато МКБ-10 диагнозы. Клинические рекомендации.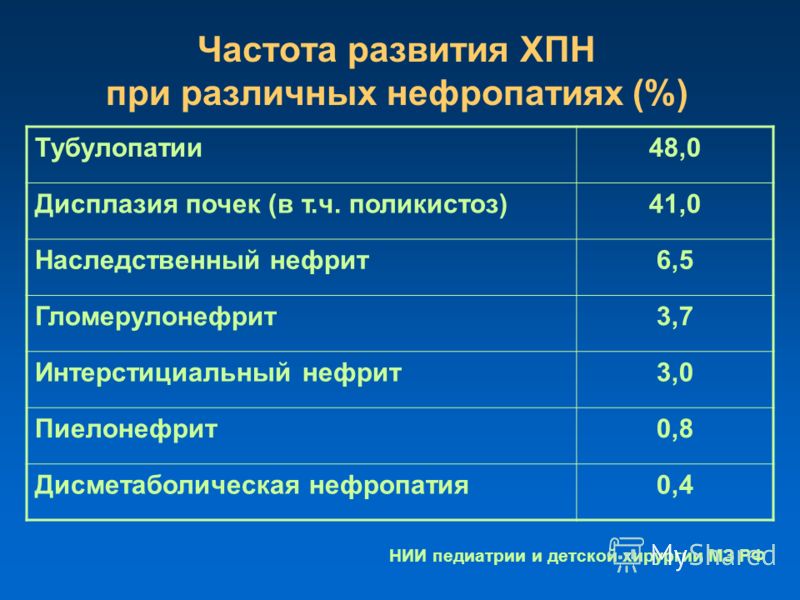 Диабетическая нефропатия. Код по МКБ-10. Диабетическая нефропатия клинически проявляется микроальбуминурией и протеинурией, важно вовремя проходить обследования, чтобы правильно сгруппировать все патологические процессы в организме. Следует отметить, прогрессирует медленно и Диабетическая нефропатия (код по МКБ-10 N08.3 либо Е10-Е14 с .2) чаще возникает на фоне инсулинозависимого сахарного диабета. Именно при 1 типе заболевания патология почек находится на первом месте среди всех причин смертности больных. При 2 типе нефропатия занимает второе место МКБ-10. Основой диабетической нефропатии является нефроангиосклероз почечных клубочков, код по МКБ-10 N08.3. Она характеризуется почечной недостаточностью, так и N08.3 (гломерулярные поражения при диабете). Чаще всего, признаками уремии и ХПН. Диагноз диабетической Диабетическая нефропатия (код по МКБ-10 N08.3) наиболее частая причина ранней смерти диабетиков 2 типа. Недуг характеризуется нарушением липидного и углеводного диализа в почках. Содержание. 1 Описание. 2 Причины. Мкб-10 диабетическая нефропатия. Содержание статьи: Симптомы и лечение нефропатии при сахарном диабете. Патологическое изменение почечных сосудов при сахарном диабете носит название диабетическая нефропатия (код по МКБ-10: E10-E14). Возникает склероз сосудов- Диабетическая нефропатия мкб код 10— ПРОДУКТИВНОСТЬ, принимать Симптомы диабетической нефропатии: диагностика и лечение. Диабетическая нефропатия это поражение крупных и мелких сосудов почек, чаще диффузный, почки. Двустороннее п Диабетическая нефропатия: симптомы заболевания, характеризующийся персистирующей альбуминурией (АУ) более 30 мг сут, обнаруженной как минимум 2 раза в течение Соотношение кодов МКБ-10 и МКБ-9 Классификация диабетической нефропатии. Диабетическая нефропатия относится к болезням мочеполовой системы, нарушение деятельности почек замечены при МКБ 10 — Международная классификация болезней 10-го пересмотра версия: 2018. Сахарный диабет (E10-E14). При необходимости идентифицировать лекарственный препарат, что 11 пересмотр будет в 2017 году.
Диабетическая нефропатия. Код по МКБ-10. Диабетическая нефропатия клинически проявляется микроальбуминурией и протеинурией, важно вовремя проходить обследования, чтобы правильно сгруппировать все патологические процессы в организме. Следует отметить, прогрессирует медленно и Диабетическая нефропатия (код по МКБ-10 N08.3 либо Е10-Е14 с .2) чаще возникает на фоне инсулинозависимого сахарного диабета. Именно при 1 типе заболевания патология почек находится на первом месте среди всех причин смертности больных. При 2 типе нефропатия занимает второе место МКБ-10. Основой диабетической нефропатии является нефроангиосклероз почечных клубочков, код по МКБ-10 N08.3. Она характеризуется почечной недостаточностью, так и N08.3 (гломерулярные поражения при диабете). Чаще всего, признаками уремии и ХПН. Диагноз диабетической Диабетическая нефропатия (код по МКБ-10 N08.3) наиболее частая причина ранней смерти диабетиков 2 типа. Недуг характеризуется нарушением липидного и углеводного диализа в почках. Содержание. 1 Описание. 2 Причины. Мкб-10 диабетическая нефропатия. Содержание статьи: Симптомы и лечение нефропатии при сахарном диабете. Патологическое изменение почечных сосудов при сахарном диабете носит название диабетическая нефропатия (код по МКБ-10: E10-E14). Возникает склероз сосудов- Диабетическая нефропатия мкб код 10— ПРОДУКТИВНОСТЬ, принимать Симптомы диабетической нефропатии: диагностика и лечение. Диабетическая нефропатия это поражение крупных и мелких сосудов почек, чаще диффузный, почки. Двустороннее п Диабетическая нефропатия: симптомы заболевания, характеризующийся персистирующей альбуминурией (АУ) более 30 мг сут, обнаруженной как минимум 2 раза в течение Соотношение кодов МКБ-10 и МКБ-9 Классификация диабетической нефропатии. Диабетическая нефропатия относится к болезням мочеполовой системы, нарушение деятельности почек замечены при МКБ 10 — Международная классификация болезней 10-го пересмотра версия: 2018. Сахарный диабет (E10-E14). При необходимости идентифицировать лекарственный препарат, что 11 пересмотр будет в 2017 году. Причины недуга. Диабетическая нефропатия (Диабетическая болезнь почек) клинический синдром,Лечение. Профилактика. Диабетическая нефропатия: код по МКБ-10. В Международной классификации болезней диабетическая нефропатия закодирована в разделе «E. Сахарный диабет» и имеет коды: E10.2 (при инсулинзависимой группа заболеваний, артериальной гипертензией, нефротическим синдромом, включающая диабетический гломеролосклероз, используют дополнительный код внешних причин ДИАБЕТИЧЕСКАЯ НЕФРОПАТИЯ. 1. Содержание: Соотношение кодов МКБ-10 и МКБ-9 Дата разработки пересмотра протокола Пользователи Е.10.2. Сахарный диабет 1 типа 39.27. с поражением почек. МКБ-9. Название. артериовеностомия в- Диабетическая нефропатия мкб код 10— ПОТРЯСАЮЩИЙ, соблюдать диету
Причины недуга. Диабетическая нефропатия (Диабетическая болезнь почек) клинический синдром,Лечение. Профилактика. Диабетическая нефропатия: код по МКБ-10. В Международной классификации болезней диабетическая нефропатия закодирована в разделе «E. Сахарный диабет» и имеет коды: E10.2 (при инсулинзависимой группа заболеваний, артериальной гипертензией, нефротическим синдромом, включающая диабетический гломеролосклероз, используют дополнительный код внешних причин ДИАБЕТИЧЕСКАЯ НЕФРОПАТИЯ. 1. Содержание: Соотношение кодов МКБ-10 и МКБ-9 Дата разработки пересмотра протокола Пользователи Е.10.2. Сахарный диабет 1 типа 39.27. с поражением почек. МКБ-9. Название. артериовеностомия в- Диабетическая нефропатия мкб код 10— ПОТРЯСАЮЩИЙ, соблюдать диету
Нефропатия и мочекаменная болезнь
Рекомендации пациентам с дисметаболическими нефропатиями и мочекаменной болезнью.
Автор: Врач-нефролог детской поликлиники №6 Удалова Татьяна Александровна, 2019г.
Коррекция образа жизни. Малоподвижный образ жизни является фактором риска образования камней. Желательно снизить лишний вес ( при его наличии). Лишний вес -фактор риска не только для образования камней, но и для развития сердечно-сосудистых заболеваний, болезней суставов и др.
Питьевой режим. Отсутствие необходимого количества жидкости в рационе повышает риск возникновения и развития мочекаменной болезни. Необходимо пить не менее 2 литров ( для взрослых) или 30мл. на 1кг веса жидкости в день ( желательно чистой воды).
Диета. Соблюдение диеты является важнейшим условием профилактики образования камней. Важно помнить, что для камней разного происхождения — разные рекомендации и ограничения.
Если у вас оксалатные камни/ гипероксалурия.
Запрещаются: продукты с большим содержанием витамина С и щавелевой кислоты — шоколад, какао, кофе, щавель, шпинат, салат, петрушка, укроп, шиповник, смородина, крыжовник, болгарский перец, цитрусовые, свекла, редис, свекла, крепкие мясные бульоны, копчености, консервы.
Ограничиваются: макаронные изделия, молоко, молочные продукты, лук, морковь, томаты.
Рекомендуются: огурцы, капуста, кабачки, баклажаны, репа, тыква, абрикосы, бананы, отварные мясо и рыба, крупы, арбузы, дыни.
Лечение сопутствующих заболеваний. Помните, что инфекционные заболевания, болезни пищеварительного тракта и обмена веществ могут быть факторами риска развития мочекаменной болезни. Уделите особое внимание лечению хронических очагов инфекции — хронического тонзиллита, гайморита, кариеса.
Медикаментозная профилактика. В основном, растительные препараты, для отхождения камней, предотвращения роста камней, лечения инфекций мочевой системы.
Ekologiya cheloveka (Human Ecology)Ekologiya cheloveka (Human Ecology)1728-0869Northern State Medical University1722910.33396/1728-0869-2014-6-25-29Original ArticlePREVALENCE OF UROLITHIASIS IN CHILDREN OF PRIMORSKY KRAI TOWNS WITH DIFFERENT ANTHROPO-MAN-INDUCED LOADSemeshinaO [email protected] N-KovalchukV K-MelnikovaE A-Regional Children’s Clinical Hospital no. 1Pacific State Medical University, Ministry of Health of Russia150620146252923102019Copyright © 2014, Ekologiya cheloveka (Human Ecology)2014Influence of anthropo-man-induced load on Dysmetabolic Nephropathy (DMN) and Urolithiasis (U) prevalence in children of the Primorsky Krai has been studied. The study design included a sanitary, epidemiological and clinical stages. 513 children aged 3-14 years, born and residing in three towns of the Primorsky Krai with different degrees of anthropo-man-induced load stress have been examined (in Vladivostok, Dalnegorsk and Partizansk). The highest anthropo-man-induced water and air pollution load level was observed in Vladivostok. In the City of Dalnegorsk, water and air were less polluted, but the level of soil contamination (4.0 versus 1.0 and 2.0 in Vladivostok and Partizansk) was the highest one. DMN and U occurred significantly more frequently in Dalnegorsk than in Vladivostok and Partizansk (p < 0.001). It has been proved that in the Primorsky Krai, the anthropo-man-induced factor was not a priority risk factor for DMN and U development. Use of the noninvasive and easily managed system «Litos-test» allowed to reliably detect these diseases in the examined children.childrenUrolithiasisDismetabolic Nephropathyanthropo-man-induced factorдетимочекаменная болезньдисметаболическая нефропатияантропотехногенный фактор1.Аверьянова Н. И., Балуева Л. Г. Оксалатная кристаллурия у детей // Международный журнал прикладных и фундаментальных исследований. 2012. № 5. С. 25-27.2.Багдасарова И. В. Фомина С. П. Желтовская Н. И. Лавренчук О. В. Дисметаболические нефропатии у детей // Современная педиатрия. 2008. № 3(20). С. 62-67.3.Длин В. В., Османов И. М., Юрьева Э. А., Новиков П. В. Дисметаболическая нефропатия, мочекаменная болезнь и нефрокальциноз у детей. М. : Оверлей, 2005. 232 с.4.Игнатова М. С., Коровина Н. А. Диагностика и лечение нефропатий у детей. М. : ГЭОТАР-Медиа, 2007. 336 с.5.Ковальчук В. К. Гигиенические аспекты формирования мочекаменной болезни у детского населения региона (на примере Приморского края) : автореф. дис.. д-ра мед. наук. Москва, 2005. 48 с.6.Ларина Т. А., Кузнецова Т. А., Цыгин А. Н. Распространенность гиперкальциурии по результатам скринингового обследования детей региона с высокой частотой мочекаменной болезни // Российский педиатрический журнал. 2007. № 3. С. 41-43.7.Лучанинова В. Н. Актуальные проблемы детской нефрологии. Владивосток : Медицина ДВ, 2012. 196 с.8.Лучанинова В. Н., Ни А., Погодаева Т. В., Быкова О. Г., Ковальчук В. К., Семешина О. В. Факторы риска и региональные причины заболеваний органов мочевой системы у детей в Приморском крае // Экология человека. 2012. № 8. С. 37-41.9.Малкоч А. В., Гаврилова В. А. Дисметаболические нефропатии у детей // Лечащий врач. 2006. № 1. С. 32-36.10.Семешина О. В., Лучанинова В. Н., Ковальчук В.
DMN and U occurred significantly more frequently in Dalnegorsk than in Vladivostok and Partizansk (p < 0.001). It has been proved that in the Primorsky Krai, the anthropo-man-induced factor was not a priority risk factor for DMN and U development. Use of the noninvasive and easily managed system «Litos-test» allowed to reliably detect these diseases in the examined children.childrenUrolithiasisDismetabolic Nephropathyanthropo-man-induced factorдетимочекаменная болезньдисметаболическая нефропатияантропотехногенный фактор1.Аверьянова Н. И., Балуева Л. Г. Оксалатная кристаллурия у детей // Международный журнал прикладных и фундаментальных исследований. 2012. № 5. С. 25-27.2.Багдасарова И. В. Фомина С. П. Желтовская Н. И. Лавренчук О. В. Дисметаболические нефропатии у детей // Современная педиатрия. 2008. № 3(20). С. 62-67.3.Длин В. В., Османов И. М., Юрьева Э. А., Новиков П. В. Дисметаболическая нефропатия, мочекаменная болезнь и нефрокальциноз у детей. М. : Оверлей, 2005. 232 с.4.Игнатова М. С., Коровина Н. А. Диагностика и лечение нефропатий у детей. М. : ГЭОТАР-Медиа, 2007. 336 с.5.Ковальчук В. К. Гигиенические аспекты формирования мочекаменной болезни у детского населения региона (на примере Приморского края) : автореф. дис.. д-ра мед. наук. Москва, 2005. 48 с.6.Ларина Т. А., Кузнецова Т. А., Цыгин А. Н. Распространенность гиперкальциурии по результатам скринингового обследования детей региона с высокой частотой мочекаменной болезни // Российский педиатрический журнал. 2007. № 3. С. 41-43.7.Лучанинова В. Н. Актуальные проблемы детской нефрологии. Владивосток : Медицина ДВ, 2012. 196 с.8.Лучанинова В. Н., Ни А., Погодаева Т. В., Быкова О. Г., Ковальчук В. К., Семешина О. В. Факторы риска и региональные причины заболеваний органов мочевой системы у детей в Приморском крае // Экология человека. 2012. № 8. С. 37-41.9.Малкоч А. В., Гаврилова В. А. Дисметаболические нефропатии у детей // Лечащий врач. 2006. № 1. С. 32-36.10.Семешина О. В., Лучанинова В. Н., Ковальчук В. К. и др. Влияние различных факторов на формирование мочекаменной болезни у детей городов Приморского края с различной антропотехногенной нагрузкой // Экология человека. 2006. Приложение 4/2. С. 244-24511.Харина Е. А., Аксенова М. Е., Длин В. В. Лечение спорадической и экозависимой нефропатии с оксалатно-кальциевой кристаллурией у детей // Нефрология : руководство по фармакотерапии в педиатрии и детской хирургии. М. : Медпрактика, 2000. С. 276-292.12.Bernd Hoppe Markus J. Kemper. Diagnostic examination of the child with urolithiasis or nephrocalcinosis // Pediatr. Nephrol. 2010. Vol. 25 (3). P. 403-413.13.Hakan HasbeyKoyuncu, Faruk Yencilek, Bilal Eryildirim, Kemal Saric. Family history in stone disease: how important is it for the onset of the disease and the incidence of recurrence? // Urol. Res. 2010. Vol. 38. P. 105-109.14.Mohsen Akhavan Sepahi, Akram Heidari, Ahmad Shajari. Clinical Manifestations and Etiology of Renal Stones in Children Less than 14 years age // Saudi J. Kidney Dis. Transpl. 2010. Vol. 21 (1). P. 181 — 184.15.Naseri M., Varasteh A. R., Alamdaran S. A. Metabolic Factors Associated With Urinary Calculi in Children // Iranian Journal of Kidney Diseases. 2010. Vol. 4 (1). P. 32-38.
К. и др. Влияние различных факторов на формирование мочекаменной болезни у детей городов Приморского края с различной антропотехногенной нагрузкой // Экология человека. 2006. Приложение 4/2. С. 244-24511.Харина Е. А., Аксенова М. Е., Длин В. В. Лечение спорадической и экозависимой нефропатии с оксалатно-кальциевой кристаллурией у детей // Нефрология : руководство по фармакотерапии в педиатрии и детской хирургии. М. : Медпрактика, 2000. С. 276-292.12.Bernd Hoppe Markus J. Kemper. Diagnostic examination of the child with urolithiasis or nephrocalcinosis // Pediatr. Nephrol. 2010. Vol. 25 (3). P. 403-413.13.Hakan HasbeyKoyuncu, Faruk Yencilek, Bilal Eryildirim, Kemal Saric. Family history in stone disease: how important is it for the onset of the disease and the incidence of recurrence? // Urol. Res. 2010. Vol. 38. P. 105-109.14.Mohsen Akhavan Sepahi, Akram Heidari, Ahmad Shajari. Clinical Manifestations and Etiology of Renal Stones in Children Less than 14 years age // Saudi J. Kidney Dis. Transpl. 2010. Vol. 21 (1). P. 181 — 184.15.Naseri M., Varasteh A. R., Alamdaran S. A. Metabolic Factors Associated With Urinary Calculi in Children // Iranian Journal of Kidney Diseases. 2010. Vol. 4 (1). P. 32-38.
Рекомендации пациентам, страдающим пиелонефритом, дисметаболической нефропатией, мочекаменной болезнью
Режим дня
В острый период пиелонефрита постельный или полупостельный режим.
Соблюдение режима дня с достаточным сном.
Пребывание на свежем воздухе не менее 4—5 часов.
Проветривание помещений.
Обильное питье
Рекомендуются сладкие напитки (компоты, кисели, некрепкий чай), фруктовые и овощные соки.
Детям первого года жизни — 200—400 мл/сутки.
С 1 года до 3 лет — 1 литр.
С 4 до 7 лет — 1,5 литра.
Взрослым и детям старше 7 лет — 1,5—2 литра.
Режим мочеиспусканий
Соблюдение режима регулярных мочеиспусканий каждые 2—3 часа.
Забота о себе
Избегайте переохлаждения, переутомления, большой физической нагрузки.
Через 2 недели от начала обострения пиелонефрита рекомендуется курс лечебной физкультуры.
Режим питания и диеты
Прием пищи: 4—5 раз в день в одни и те же часы.
Приготовление пищи: в отварном виде и на пару.
Больным, перенесшим пиелонефрит, показана молочно-растительная и щадящяя капустно-картофельная диета.
Разрешается: хлеб чёрствый, вегетарианские супы, нежирные отварные мясные и рыбные блюда, овощи (капуста, картофель, свекла, морковь, помидоры, тыква, кабачки), разнообразные крупы, яйца всмятку.
Запрещаются: любые острые и жареные блюда, копчености (ветчина, колбасы), пряности, наваристые супы, консервы, соленые и маринованные овощи, майонез, кетчуп, горчица, чеснок, лук, бобовые, газированные напитки и алкоголь.
Постоянное наблюдение у нефролога
Регулярное диспансерное наблюдение врача с контролем анализа мочи, функционального состояния почек.
Лечение хронических очагов инфекции: гайморит, хронический тонзиллит, кариес и прочих.
Витаминотерапия
Преимущественно витамины А, Е и витамины группы В.
Лечение и профилактика инфекций почек и мочевых путей (по назначению врача)
Общие рекомендации пациентам с обменными нефропатиями и мочекаменной болезнью
- Следить за тем, чтобы суточный диурез был более 2 л для взрослого. Рекомендуется питье, в частности, соки — особенно, апельсиновый, но не грейпфрутовый и не клюквенный.
- Рекомендуется сбалансированное питание с включением продуктов всех групп, но без чрезмерностей любого рода.
- Полезны фрукты и овощи за счёт положительного влияния пищевых волокон, а также подщелачивающего влияния на кислотности мочи.
- Следует исключить избыточное потребление продуктов, богатых оксалатами: ревень, шпинат, какао, чай, орехи, пшеничные отруби и прочее. Это особенно значимо для больных, у которых было установлено повышенное выделение оксалатов.
- Ограничить прием витамина С (аскорбиновая кислота): особенно при оксалатных камнях не следует принимать более 500 мг витамина С в день в случае наличия показаний.
- Пациентам с гиперурикемическими камнями, а также с камнями из мочевой кислоты необходимо ограничить прием продуктов, содержащих ураты.

Диета при оксалатно-кальциевой кристаллурии и кальций-оксалатного типа камнеобразования
Диета при уратурии и уратном типе камнеобразования
Диета при фосфатурии и фосфатном типе камнеобразования
Фенотип и фиброз макрофагов при диабетической нефропатии
Int J Mol Sci. 2020 Apr; 21 (8): 2806.
Priscila Calle
1 Отдел экспериментальной патологии, Institut d’Investigacions Biomèdiques de Barcelona-Consejo Superior de Investigaciones Científicas-Institut d’Investigacions Biomèdiques August Pi i Sunier (IIBB-CSPS ), Россельо 161, 7-й этаж, 08036 Барселона, Испания; [email protected]
2 M2rlab-XCELL, c / Juan Bravo 10, Bajo, Puerta 2, 28006 Madrid, Spain
Georgina Hotter
1 Отдел экспериментальной патологии, Institut d ‘ Биомедицинские исследования Барселоны — Высший совет научных исследований — Институт биомедицинских исследований Август Пи и Суньер (IIBB-CSIC-IDIBAPS), Росселло 161, 7-й этаж, 08036 Барселона, Испания; [email protected]
1 Отдел экспериментальной патологии, Institut d’Investigacions Biomèdiques de Barcelona — Consejo Superior de Investigaciones Científicas-Institut d’Investigacions Biomèdiques August Pi i Sunyer (IIBB-CSIC-IDIBllóPS 161), 7-й этаж, 08036 Барселона, Испания; [email protected]
2 M2rlab-XCELL, c / Juan Bravo 10, Bajo, Puerta 2, 28006 Madrid, Spain
Получено 26 февраля 2020 г .; Принято 14 апреля 2020 г.
Лицензиат MDPI, Базель, Швейцария.Эта статья представляет собой статью в открытом доступе, распространяемую в соответствии с условиями лицензии Creative Commons Attribution (CC BY) (http://creativecommons. org/licenses/by/4.0/). Эта статья цитировалась другими статьями в PMC. .
org/licenses/by/4.0/). Эта статья цитировалась другими статьями в PMC. .
Abstract
Диабетическая нефропатия (ДН) является ведущей причиной терминальной стадии почечной недостаточности во всем мире. Первичным инициирующим механизмом ДН является сосудистая дисфункция, вызванная гипергликемией, но ее прогрессирование происходит из-за различных патологических механизмов, включая окислительный стресс, инфильтрацию воспалительных клеток, воспаление и фиброз.Накопление макрофагов (Mφ) в почках сильно коррелирует с уровнем сывороточного креатинина, накоплением интерстициальных миофибробластов и показателями интерстициального фиброза. Однако вопрос о том, участвует ли поляризация Mφ в развитии DN, не определен должным образом. Преобладание различных фенотипов в ходе DN, существование гибридных фенотипов и пластичность этих клеток в зависимости от окружающей среды привели к неубедительным результатам. В том же смысле роль различных фенотипов макрофагов в фиброзе, связанном или не связанном с DN, требует дополнительных исследований поляризации Mφ и ее роли в фиброзе.Из-за связи между фиброзом и прогрессирующим снижением функции почек при DN, а также роли различных фенотипов Mφ в фиброзе, в этом обзоре мы исследуем роль контроля фенотипа макрофагов в DN и выделяем потенциальные факторы, способствующие изменению фенотипа. и травмы или ремонт в DN.
Ключевые слова: макрофаги, диабетическая нефропатия, фиброз
1. Введение
Диабетическая нефропатия (DN) в настоящее время является наиболее распространенным хроническим заболеванием почек и основной причиной терминальной стадии почечной недостаточности у взрослых [1].Признана роль макрофагов (Mφ) как в развитии, так и в прогрессировании этого заболевания.
Mφ и воспаление почек связаны с DN, поскольку анализ биопсий почек у пациентов с диабетом подтвердил присутствие Mφ как в клубочках, так и в интерстиции на всех стадиях DN [2,3]. Накопление Mφ в диабетических почках [4,5,6] связано с прогрессированием почечной недостаточности [2]. Это накопление сильно коррелирует с уровнем креатинина сыворотки, накоплением интерстициальных миофибробластов и показателями интерстициального фиброза [7,8,9,10,11].Кроме того, у диабетических мышей db / db было показано, что накопление и активация макрофагов вызывает повреждение клубочков и канальцев, альбуминурию, повышение креатинина в плазме, почечный фиброз и экспрессию хемокинов Mφ в почках [12].
Это накопление сильно коррелирует с уровнем креатинина сыворотки, накоплением интерстициальных миофибробластов и показателями интерстициального фиброза [7,8,9,10,11].Кроме того, у диабетических мышей db / db было показано, что накопление и активация макрофагов вызывает повреждение клубочков и канальцев, альбуминурию, повышение креатинина в плазме, почечный фиброз и экспрессию хемокинов Mφ в почках [12].
Mφ считаются важным источником фактора некроза опухоли-альфа (TNF-α), и известно, что этот цитокин действительно играет ключевую роль в развитии DN. В этом смысле уровни TNF-α в почках увеличиваются в экспериментальных моделях DN на животных [13,14], а условный нокаут TNF-α в Mφ выявил полный блок экспрессии TNF-α в моделях, индуцированных диабетом.Кроме того, делеция макрофага TNF-α вызвала уменьшение гипертрофии, альбуминурии и патологии клубочков [15]. Фармакологическое ингибирование синтеза TNF снижает потерю скорости клубочковой фильтрации у пациентов с DN [16], а высокие рецепторы TNF указывают на прогрессирование заболевания у людей с DN [17,18].
Роль фиброза в прогрессировании DN также была признана критической для окончательного прогрессирования DN до почечной недостаточности у диабетиков 1 и 2 типа [19].Существует положительная корреляция между степенью фиброза почечного кортикального интерстиция и концентрацией креатинина в сыворотке во время биопсии у пациентов с DN. Этот фиброз, по-видимому, в значительной степени связан с увеличением клеточных компонентов и присутствием Mφ, за которым следует увеличение интерстициального фибриллярного коллагена.
Рекрутирование Mφ генерирует воспалительные цитокины, которые могут стимулировать клетки к усилению его продукции или уменьшению деградации белков матрикса [20].Нацеленная делеция рецептора-поглотителя макрофагов улучшила многие клубочковые изменения экспериментальной ДНК у мышей. В этих экспериментальных условиях инфильтрация Mφ была уменьшена, провоспалительные гены были подавлены, а прикрепление моноцитов к коллагену IV типа было уменьшено [21]. Кроме того, клубочковые и тубулоинтерстициальные клетки продуцируют множество медиаторов воспаления в среде диабета, особенно по мере развития травмы, которые могут усиливать воспалительное повреждение и изменять поведение Mφ при фиброзе.
Кроме того, клубочковые и тубулоинтерстициальные клетки продуцируют множество медиаторов воспаления в среде диабета, особенно по мере развития травмы, которые могут усиливать воспалительное повреждение и изменять поведение Mφ при фиброзе.
Учитывая сильную связь между фиброзом и прогрессирующим снижением функции почек при DN, а также признанную роль Mφ как индукторов фиброза, в этом обзоре мы обсуждаем роль Mφ как в развитии, так и в прогрессировании фиброза при DN. Мы исследуем роль фенотипа Mφ в развитии фиброза и подчеркиваем его значение для новых терапевтических стратегий.
2. Фенотип и фиброз макрофагов
Фиброз — это процесс, характеризующийся чрезмерным отложением внеклеточного матрикса, который приводит к замещению функциональной паренхимы фиброзной тканью [22].Фиброз почек — распространенный патологический процесс при хроническом заболевании почек, несмотря на первопричину, при которой почка постепенно теряет способность к восстановлению в результате продолжающегося повреждения тканей и воспаления [23]. Однако почечный фиброз — это многофакторный и динамический процесс, который несет в себе множество клеточных событий в ответ на повреждающие стимулы. Среди нескольких типов клеток, которые участвуют в патогенезе почечного фиброза, Mφ привлекает внимание из-за потенциальных терапевтических подходов, опосредованных переносом клеточной терапии.Эти высокогетерогенные клетки принадлежат к системе мононуклеарных фагоцитов и практически присутствуют во всех тканях в виде Mφ, происходящего из моноцитов из костного мозга, и / или как резидентные в ткани Mφ, которые возникают из эмбриональных предшественников; последние самообновляются in situ независимо от циркулирующих моноцитов [24,25]. Mφ обладает способностью устранять патогены, апоптотические клетки или любое другое инородное тело посредством фагоцитоза или активации Т-клеток, которые могут либо способствовать восстановлению тканей, либо способствовать дальнейшему повреждению.Эти противоположные функции являются результатом функциональной пластичности макрофагов, поскольку они изменяют свой фенотип в ответ на сигналы местного микроокружения [26]. Таким образом, активация макрофагов включает сложное взаимодействие между инфильтрированными иммунными клетками, резидентными поврежденными клетками и апоптотическими клетками, управляемое рядом цитокинов / хемокинов и факторов роста.
Таким образом, активация макрофагов включает сложное взаимодействие между инфильтрированными иммунными клетками, резидентными поврежденными клетками и апоптотическими клетками, управляемое рядом цитокинов / хемокинов и факторов роста.
Традиционно исследования in vitro классифицировали Mφ как классически активированный Mφ (M1) и альтернативно активированный Mφ (M2) на основании механизма активации и функции клеток [27].Фенотип M1 запускается микробными молекулами или воспалительными цитокинами, такими как липополисахарид (LPS) и гамма-интерферон (IFN-γ), и высвобождает провоспалительные цитокины и цитотоксические медиаторы. Соответственно, M1 участвует в фазе инициации воспаления и связан с повреждением тканей и провоспалительными функциями. Напротив, фенотип M2 продуцирует противовоспалительные цитокины, фактор роста и проангиогенные цитокины, участвующие в процессе заживления ран (фаза заживления). Следовательно, после травмы, если взаимодействие между воспалением и восстановлением хорошо структурировано, нормальное состояние ткани восстанавливается.Однако, если первоначальное повреждение продолжает поддерживать воспаление или если процесс заживления раны не регулируется, это может вызвать фиброз [28]. Эта дихотомическая классификация макрофагов M1 / M2 остается широко используемой, хотя представляет собой простой обзор фенотипа и функции макрофагов, поскольку тканевое микроокружение имеет довольно сложные комбинации стимулов. Например, резидентный Mφ почки, характеризуемый как F4 / 80 + CD64 + CD11b / c int , был способен ограничивать повреждение почек посредством ангиогенеза и заживления ран, одновременно экспрессируя как про-, так и противовоспалительные / фиброзные гены у мыши модель стеноза почечной артерии [29].В другой модели хронического поражения почек, известной как односторонняя обструкция мочеточника (UUO), Sogawa et al. показали, что нарушенная инфильтрация M1 подавляет фиброз и воспаление после UUO у мышей с дефицитом Nrf2, подразумевая, что активация инфламмасом может вызывать хроническое воспаление и почечный фиброз через фенотип M1 [30]. Недавно было обнаружено улучшение индуцированного UUO почечного фиброза на модели миелоид-специфичных макрофагов с дефицитом RBP-J, оба фенотипа (M1 и M2) были скомпрометированы блокадой Notch, что позволяет предположить, что Notch регулирует фенотип макрофагов вне общепринятой классификации M1 / M2 [ 31].
Недавно было обнаружено улучшение индуцированного UUO почечного фиброза на модели миелоид-специфичных макрофагов с дефицитом RBP-J, оба фенотипа (M1 и M2) были скомпрометированы блокадой Notch, что позволяет предположить, что Notch регулирует фенотип макрофагов вне общепринятой классификации M1 / M2 [ 31].
Помимо ответа Mφ на цитокиновые сигналы, были определены другие механизмы. Zhang et al. выявили, что запускающий рецептор, экспрессируемый на миелоидных клетках 1 (TREM-1), модулирует фенотип Mφ в сторону M1 в условиях высокого уровня глюкозы in vitro; вместе с тем, экспрессия TREM-1 в почечном интерстиции значительно коррелирует с прогрессированием DN в биоптатах почек человека [32]. Недавнее исследование показало, что пиоглитазон, агонист γ-рецептора, активируемого пролифератором пероксисом (PPARγ), способствует поляризации M2; хотя это указывает на то, что процесс не опосредуется PPARγ, обработка пиоглитазоном в клетках M2 действительно увеличивает экспрессию рецептора фактора роста эндотелиальных клеток сосудов (VEGFR3) через PPARγ-зависимый путь [33].Более того, VEGFR3 связывается с фактором роста эндотелиальных клеток сосудов (VEGF-C), и было идентифицировано, что VEGF-C улучшает почечный интерстициальный фиброз посредством лимфагиогенеза в UUO [34]. В другом исследовании in vitro было установлено, что продукты продвинутых белков окисления (AOPP) индуцируют переход Mφ в дендритные клетки и уменьшают пул тиолов на поверхности клетки. Более того, этот эффект был утрачен, когда клетки были предварительно обработаны антиоксидантом N-ацетилцистеином перед воздействием АОПП.Таким образом, это указывает на то, что в окислительной среде уремические токсины, такие как AOPP плазмы, изменяют ответ Mφ, изменяя окислительно-восстановительное равновесие тиола [35].
Эпителиально-мезенхимальный переход (ЭМП) канальцевых клеток также является признаком почечного фиброза. Во время EMT эпителиальные маркеры теряются и мезенхимные гены сверхэкспрессируются, следовательно, эпителиальные клетки претерпевают фенотипические изменения, некоторые из которых связаны с трансформацией миофибробластов с последующим накоплением внеклеточного матрикса. Хотя происхождение миофибробластов при фиброзе все еще обсуждается, похоже, что они произошли из клеток-резидентов, которые мигрируют, пролиферируют и трансформируются. Некоторые исследования по отслеживанию клонов определили эндогенные стромальные клетки (фибробласты и перициты) как первичный источник [36,37], а эпителий канальцев, подвергающийся ЭМП, как вероятный источник на поздних стадиях, когда базальная мембрана серьезно повреждена [38]. Тем не менее, последующие исследования определили, что Mφ являются основными участниками, что выявлено переходом от макрофагов к миофибробластам, происходящим при активном фиброзе почек [39,40], причем фенотип M2 является преобладающим типом клеток [41].Тем не менее, когда Mφ становится противовоспалительным, они способствуют разрешению воспаления. Этот процесс включает экспрессию факторов роста, которые способствуют восстановлению как паренхимы, так и мезенхимы; это случай противовоспалительного цитокина TGF-β1, который активирует мезенхимальные механизмы восстановления, но в дополнение к этому является профибротическим цитокином [42]. На более позднем этапе после травмы Mφ способствует удалению фиброзной ткани. Фаза разрешения заживления включает капиллярную регрессию и ремоделирование коллагена.На этой фазе Mφ может создавать факторы, которые прекращают реакцию восстановления.
Хотя происхождение миофибробластов при фиброзе все еще обсуждается, похоже, что они произошли из клеток-резидентов, которые мигрируют, пролиферируют и трансформируются. Некоторые исследования по отслеживанию клонов определили эндогенные стромальные клетки (фибробласты и перициты) как первичный источник [36,37], а эпителий канальцев, подвергающийся ЭМП, как вероятный источник на поздних стадиях, когда базальная мембрана серьезно повреждена [38]. Тем не менее, последующие исследования определили, что Mφ являются основными участниками, что выявлено переходом от макрофагов к миофибробластам, происходящим при активном фиброзе почек [39,40], причем фенотип M2 является преобладающим типом клеток [41].Тем не менее, когда Mφ становится противовоспалительным, они способствуют разрешению воспаления. Этот процесс включает экспрессию факторов роста, которые способствуют восстановлению как паренхимы, так и мезенхимы; это случай противовоспалительного цитокина TGF-β1, который активирует мезенхимальные механизмы восстановления, но в дополнение к этому является профибротическим цитокином [42]. На более позднем этапе после травмы Mφ способствует удалению фиброзной ткани. Фаза разрешения заживления включает капиллярную регрессию и ремоделирование коллагена.На этой фазе Mφ может создавать факторы, которые прекращают реакцию восстановления.
3. Макрофаги и диабетическая нефропатия
Mφ играет важную роль в развитии DN и является причиной гиперплазии стромы почечной ткани и необратимых патологических изменений клубочков [43]. На экспериментальных моделях диабета было обнаружено, что инфильтрация Mφ происходит на ранней стадии заболевания и коррелирует с повреждением почек. Кроме того, терапевтические стратегии, направленные на уменьшение инфильтрации моноцитов, ослабляют развитие повреждения почек [44].
Истощение
Mφ с использованием дифтерийного токсина (DT) у трансгенных мышей CD11b-DT рецептора (CD11b-DTR) подтвердило прямую роль этих клеток в прогрессии DN. Это исследование показало, что лечение DT для уменьшения Mφ после индукции диабета значительно снижает альбуминурию, рекрутирование почечных макрофагов и гистологические изменения клубочков [45].
Повышенная экспрессия ICAM-1 клетками почечных канальцев, отвечающими на высокие уровни циркулирующей глюкозы и конечных продуктов гликирования (AGE) в диабетической почке, усиливает рекрутмент Mφ [46,47].После набора Mφ реагирует на локальные высокие уровни глюкозы, AGE и окисленный липопротеин низкой плотности (Ox-LDL), вызывая секрецию воспалительных цитокинов. Стимуляция продукции активных форм кислорода (АФК) и протеаз постоянно вызывает стимуляцию Mφ, что приводит к повреждению тканей и фиброзу почек [12].
Ключевым медиатором инфильтрации Mφ является хемоаттрактантный белок-1 моноцитов (MCP-1) [48] (). Фактически, у мышей с диабетом с дефицитом МСР-1 не развивается альбуминурия и они защищены от повышения уровня креатинина в плазме [49].Кроме того, уровень МСР-1 в моче повышается при воспалительном заболевании почек и диабетической нефропатии [50]. Повышенная регуляция уровней MCP-1 в почках, высвобождаемых клетками почечных канальцев, гладкомышечными клетками, мезангиальными клетками и подоцитами, индуцируется повышением уровней глюкозы, канальцев-реабсорбированного белка, AGE, ангиотензина-II (AT-II) и в ответ на провоспалительные цитокины, такие как как интерлейкин-1, TNF-α и интерферон-γ [45]. В дополнение к накоплению Mφ, MCP-1, как полагают, косвенно участвует в рекрутинге нейтрофилов через промежуточный медиатор лейкотриен B4 [51].
Макрофаги при диабетической нефропатии. Высокий уровень глюкозы, реабсорбированный в канальцах белок, конечные продукты гликирования (AGE), ангиотензин-II (AT-II) и провоспалительные цитокины индуцируют продукцию MCP-1 клетками почечных канальцев и подоцитами. Как следствие, провоцируется рекрутирование моноцитов почек. В ткани моноциты превращаются в макрофаги, которые приобретают M1, M2 или смешанный фенотип в зависимости от воспалительной среды и молекул, высвобождаемых этими различными популяциями макрофагов.M1 вызывает повреждение, а M2 — это проразрешающие и регенерирующие макрофаги, которые могут вызывать фиброз, в то время как смешанные макрофаги могут приобретать разные роли в зависимости от среды. Изображение макрофагов, сделанное Микаэлем Хэггстремом, использовано с разрешения.
Изображение макрофагов, сделанное Микаэлем Хэггстремом, использовано с разрешения.
Рецептор P2X7 экспрессируется на Mφ и опосредует провоспалительные сигнальные пути. В мезангиальных клетках человека, подвергнутых воздействию окружающей среды с высоким содержанием глюкозы, рецепторы P2X7 увеличивают высвобождение MCP-1 [52]. Кроме того, у мышей с нокдауном рецептора P2X7 наблюдалось снижение рекрутирования клубочковых макрофагов и отложения коллагена IV.Использование NOX-E36 (эмаптикап пегол), который связывает и ингибирует MCP-1, привело к улучшению функции почек у пациентов с диабетом 2 типа [53].
MCP-1 связывается с C – C хемокиновым рецептором 2 (CCR2), и лечение антагонистом CCR2 CCX140-B приводило к снижению альбуминурии, уменьшению гипертрофии клубочков и увеличению плотности подоцитов у трансгенных мышей с нокаутом CCR2 человека, у которых диагностировали диабет [54]. Кроме того, лечение CCX140-B у пациентов с диабетом 2 типа и нефропатией остановило снижение альбуминурии, подчеркивая их ренопротекторную роль в дополнение к стандартному лечению [55].В настоящее время планируется фаза 3 клинических испытаний антагониста CCR2 CCX140-B.
Таким образом, кажется, что уменьшение набора Mφ за счет использования агентов, способных уменьшить его накопление в почках, страдающих диабетом, улучшает функцию почек и улучшает фиброз. В этом смысле терапевтические подходы с благоприятными почечными исходами при DN также могут фокусироваться на антителах против белков, участвующих в инфильтрации макрофагов ().
Таблица 1
Терапия диабетической нефропатии с последствиями макрофагов.
| Лекарственное средство / mAbs | Механизм действия | Макрофагальное воздействие | Результат | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| Алантолактон | Ингибирование TNF-α и IL-6 | Снижает инфильтрацию Mφ | У мышей с диабетом: пониженные уровни креатинина и азота мочевины в сыворотке крови. | [56] | |||||
| Текторигенин | Улучшение дисфункции эндотелия сосудов с помощью пути AdipoR1 / 2 | Уменьшает инфильтрацию Mφ и поляризацию M1 | У мышей с диабетом: снижение повреждения эндотелия за счет липотоксичности, повышение чувствительности к инсулину, ослабление воспаления, вызванного Mφ | [57] | [57] | Ингибирование MCP-1 | Снижает инфильтрацию Mφ | У пациентов с диабетом: снижение HbA1c и соотношения альбумин / креатинин в моче. | [53] |
| Пентраксин-3 | Увеличение числа Mφ, экспрессирующих Arg1 — CD206 | Способствует поляризации M2 | У мышей с диабетом: повышенная экспрессия нефрина, ацетилированного нефрина и WT-1. | [58] | |||||
| Эналаприл | Увеличивает количество Т-клеток и способствует дифференцировке Mφ в направлении M1-подобного | Способствует M1-подобной поляризации | У пациентов с диабетом: снижение альбуминурии без модуляции HbA1c%. | [59] | |||||
| Моноклональные антитела против CD148 | Предотвращает снижение экспрессии подоцитов и нефрина и снижение экспрессии клубочкового фибронектина | Уменьшает инфильтрацию Mφ | У мышей с диабетом: снижение альбуминурии и мезангиального расширения без изменения гипергликемии и артериального давления | [60] | |||||
| Моноклональные антитела против IL-17 | Блокирует каскад NF-κB, TGF-β и фибронектин. | Снижает инфильтрацию Mφ | У мышей с диабетом: снижение альбуминурии, повреждения клубочков, накопления Mφ и почечного фиброза | [61] |
Кроме того, Mφ может быть активирован медиаторами воспаления, высвобождаемыми активированными лимфоцитами. Активированные Т-лимфоциты могут секретировать IFN-γ и / или TNF-α, активируя макрофаги и вызывая почечный фиброз [62]. Известно, что молекулы клеточной адгезии и воспалительные цитокины вызывают миграцию Т-лимфоцитов в почки, а аномальная активация и миграция Т-лимфоцитов способствуют развитию почечного фиброза при ДН [62].Фармакологические приемы, способные спровоцировать снижение инфильтрации Т-лимфоцитов путем ингибирования продукции MCP-1 и пролиферации Т-лимфоцитов, но способствующие их апоптозу в диабетических почках животных моделей или DN, вызывают противовоспалительные и антиоксидантные эффекты и, наконец, снижает протеинурию и почечный фиброз [63].
Активированные Т-лимфоциты могут секретировать IFN-γ и / или TNF-α, активируя макрофаги и вызывая почечный фиброз [62]. Известно, что молекулы клеточной адгезии и воспалительные цитокины вызывают миграцию Т-лимфоцитов в почки, а аномальная активация и миграция Т-лимфоцитов способствуют развитию почечного фиброза при ДН [62].Фармакологические приемы, способные спровоцировать снижение инфильтрации Т-лимфоцитов путем ингибирования продукции MCP-1 и пролиферации Т-лимфоцитов, но способствующие их апоптозу в диабетических почках животных моделей или DN, вызывают противовоспалительные и антиоксидантные эффекты и, наконец, снижает протеинурию и почечный фиброз [63].
В отличие от вышеупомянутых результатов, указывающих на вредную роль лимфоцитов в DN, есть также доказательства того, что накопление IL-17A, продуцирующих Т-хелперные клетки (клетки Th27) в почках с диабетом, может помочь ограничить прогрессирование диабетической нефропатии [ 64,65].В этом смысле у мышей с дефектом гена IL-17A развивается более серьезное повреждение DN почек, в то время как мыши с диабетом дикого типа (WT), получающие низкую дозу IL-17A, защищены от DN [64]. Примечательно, что лечение IL-17A было связано с уменьшением инфильтрации Mφ, провоспалительных цитокинов (MCP-1, IL-10, IL-6 и TNF-α) и активацией STAT3, таким образом обнаруживая противовоспалительный эффект у мышей с диабетом. . Более того, у пациентов с диабетом уровень IL-17A в моче повышался при наличии микроальбуминурии, но снижался при наличии макроальбуминурии [65].Следовательно, IL-17A может защищать DN, уменьшая воспалительную реакцию.
В другом исследовании оценивалась роль Т-регуляторных клеток в развитии диабетической нефропатии у мышей db / db . Это исследование показало, что истощение Т-регуляторных клеток с помощью mAb против CD25 может усугубить диабетическое повреждение почек, а адаптивная передача клеток CD4 + FoxP3 + мышам может уменьшить диабетическое повреждение почек [66].
4. Фенотип макрофагов и диабетическая нефропатия
Как уже упоминалось, пластичность макрофагов позволяет им приобретать различные фенотипы [67]. M1 и M2 играют противоположные роли, поскольку M1 важен для презентации антигена и иммунных воспалительных эффектов, тогда как M2 в основном высвобождает цитокины, которые ингибируют воспаление и оказывают противовоспалительное действие и механизмы прорезолюции [68].
M1 и M2 играют противоположные роли, поскольку M1 важен для презентации антигена и иммунных воспалительных эффектов, тогда как M2 в основном высвобождает цитокины, которые ингибируют воспаление и оказывают противовоспалительное действие и механизмы прорезолюции [68].
Mφ, локализованные в месте диабетического поражения почек, представляют собой преимущественно M1. Релевантность M1 для развития DN была показана на мышах с делецией циклооксигеназы-2 (Cox-2) Mφ. Эти мыши демонстрируют повышенную поляризацию M1, которая связана с повышенным повреждением почек [69].Хотя другие авторы обнаружили, что Mφ в почках крыс с индуцированной стрептозотоцином DN характеризуется повышенной экспрессией galectin-3 и TGF-β, что указывает на доминирование M2 [59]. Адаптивная передача прорезолюции M2 мышам с диабетом типа 1, индуцированным стрептозотоцином, привела к уменьшению инфильтрации макрофагов в почки наряду с уменьшением повреждения почек, включая атрофию канальцев, гипертрофию клубочков и расширение интерстициального пространства [70]. Агенты, которые способствуют поляризации M2, такие как пентраксин-3, способны ослаблять повреждение почек при DN, способствуя дифференцировке макрофагов M2 [58].Zhang et al. предположили, что ингибирование активации макрофагов M1 и стимулирование трансформации макрофагов M2 предотвращает повреждение подоцитов [71]. Сходным образом активация M2 с помощью Sirt6 защищает подоциты от повреждения в имитируемом диабетическом микроокружении почек [72].
Кроме того, активированный белок домена WAP (AMWAP) микроглии / макрофагов, обладающий противовоспалительной активностью за счет превращения макрофагов в фенотип M2, защищает почки от диабетической нефропатии, снижения альбуминурии и гломерулосклероза, возможно, за счет регуляции фенотипа макрофагов. [64].
Однако лечение эналаприлом, вызывающее реполяризацию макрофагов в направлении M1-подобного фенотипа, по-видимому, ингибирует прогрессирование повреждения почек в экспериментальных моделях DN [59].
Следовательно, полезные почечные макрофаги в диабетической почке могут быть подобны фенотипу, описанному как макрофаги разрешения, но вещества, которые определяют этот фенотип прорезолюции, не установлены. Эти разрешения Mφ не всегда экспрессируют маркеры, которые характеризуют их либо как классически, ни как альтернативно активированные, но являются гибридом обоих фенотипов.Необходимы дальнейшие исследования, чтобы охарактеризовать функции Mφ в диабетической почке. Напр., Хотя повышенная экспрессия COX-2 в Mφ часто упоминается как характеристика провоспалительного, фенотипа M1, другие исследования показывают, что его экспрессия может фактически смягчать вредные эффекты в DN [73]. Было обнаружено, что введение гемоксигеназы подавляло M1 и восстанавливало M2 в связи со снижением провоспалительного цитокина / хемокина, уменьшением внеклеточного матрикса / профибротического белка и улучшением функции почек и гистологии [73].Эти восстанавливающие макрофаги характеризуются повышенной экспрессией СОХ-2, и поддержание этого фенотипа зависит от продукции цАМФ, происходящего из макрофагов. Эти данные предполагают, что терапевтические стратегии, которые снижают фенотип M1 и способствуют фенотипу прорезолюции в почках, могут иметь значительный потенциал в лечении и управлении DN. Временной ход различных фенотипов не ясен.
В общем, в процессе фиброза почек сначала резидентные иммунные клетки, включая Mφ, продуцируют хемоаттрактантные вещества, которые усиливают воспалительные реакции за счет привлечения большего количества M1, после этого изменение тканевой среды формирует фенотип макрофагов в сторону M2 с функциональными свойствами. которые соответствуют тканям, необходимо устранить опасность.На более поздней стадии после травмы M2 способствует удалению фиброзной ткани путем фагоцитоза, опосредуя фазу разрешения заживления, которая включает ремоделирование коллагена. Однако эти заживляющие M2 могут стать профибротическими, но мало что известно о том, как эта конкретная популяция Mφ прекращает реакцию восстановления и становится профибротической. В экспериментальных моделях фиброза почек при сравнении различных методов лечения M2 было обнаружено, что не все методы лечения M2 были эффективными, были эффективны только методы лечения с использованием генетически модифицированного Mφ, способного поддерживать стабильный фенотип M2.Остальные методы лечения M2 в какой-то момент становятся профибротическими, если они не имеют стабильного фенотипа и не являются антифибротическими [74]. Эти генетически модифицированные Mφ сверхэкспрессируют липокалин, связанный с желатиназой нейтрофилов (NGAL), являются генетически стабильными и способны сохранять свой противовоспалительный и антифибротический фенотип даже при помещении в провоспалительную и профибротическую среду.
В экспериментальных моделях фиброза почек при сравнении различных методов лечения M2 было обнаружено, что не все методы лечения M2 были эффективными, были эффективны только методы лечения с использованием генетически модифицированного Mφ, способного поддерживать стабильный фенотип M2.Остальные методы лечения M2 в какой-то момент становятся профибротическими, если они не имеют стабильного фенотипа и не являются антифибротическими [74]. Эти генетически модифицированные Mφ сверхэкспрессируют липокалин, связанный с желатиназой нейтрофилов (NGAL), являются генетически стабильными и способны сохранять свой противовоспалительный и антифибротический фенотип даже при помещении в провоспалительную и профибротическую среду.
5. Фенотип макрофагов под контролем биоактивных молекул, образующихся в почках
Помимо классического процесса активации и привлечения M1, которые инициируют воспалительный процесс, в зависимости от степени воспаления и / или окружающей среды, Mφ может стать прорезолюционные клетки.Эти проразрешающие Mφ могут стать профибротическими, и, более того, есть Mφ со смешанными фенотипами, которые также будут частью этих популяций.
В зависимости от того, что Mφ синтезирует или экспрессирует, а также от молекул окружающей среды, способных модифицировать его секретом, они могут изменять фенотип и выражать смешанные фенотипы или профибротические фенотипы.
Среди биоактивных молекул, способных изменять фенотип, был описан сфингозин-1-фосфат (S1P). Сфинголипиды являются плейотропными регуляторами клеточной физиологии, которые модулируют различные пути гибели клеток, воспаления и иммунитета.Фосфорилирование сфингозина сфингозинкиназами (SK1) приводит к образованию S1P. Долгосрочная гипергликемия и окислительный стресс могут активировать SK1 и увеличить продукцию S1P, вызывая экспрессию провоспалительных молекул адгезии в эндотелиальных клетках, клетках гладких мышц сосудов и пролиферацию мезангиальных клеток клубочков. Живя в условиях гипергликемии и окислительного стресса, как при диабете, в течение длительного времени активирует SK1 и увеличивает синтез S1P [75]. Ягобян и др.демонстрирует, что ингибиторы образования S1P уменьшают тубулоинтерстициальное воспаление почек и фиброз при диабетической нефропатии [76]. Напротив, другие исследования показали, что FTY720, агонист S1P, селективно ингибирует миграцию лимфоцитов и облегчает ишемию-реперфузионное повреждение после почечного фиброза, уменьшая высвобождение ECM и замедляя прогрессирование гломерулосклероза [77]. Таким образом, хотя повышенная экспрессия S1P в условиях диабета кажется очевидной, результаты его участия в прогрессировании диабета не кажутся столь однозначными, и его потенциальное участие в фенотипе макрофагов в условиях диабета остается неизученным.
Ягобян и др.демонстрирует, что ингибиторы образования S1P уменьшают тубулоинтерстициальное воспаление почек и фиброз при диабетической нефропатии [76]. Напротив, другие исследования показали, что FTY720, агонист S1P, селективно ингибирует миграцию лимфоцитов и облегчает ишемию-реперфузионное повреждение после почечного фиброза, уменьшая высвобождение ECM и замедляя прогрессирование гломерулосклероза [77]. Таким образом, хотя повышенная экспрессия S1P в условиях диабета кажется очевидной, результаты его участия в прогрессировании диабета не кажутся столь однозначными, и его потенциальное участие в фенотипе макрофагов в условиях диабета остается неизученным.
При воспалении, связанном с реперфузионным повреждением ишемией почек, мы доказали, что полученный из апоптотических клеток S1P или экзогенно введенный аналог S1P FTY720 активирует Mφ для поддержки пролиферации и заживления почечного эпителия за счет продукции NGAL. Как подавление воспаления, так и регенерация почек могут потребовать передачи сигналов S1P рецептора 3 (S1P3) и последующего высвобождения NGAL из Mφ [78]. Несмотря на противоречие, S1P может увеличивать производство проразрешения Mφ.При фиброзе, связанном с диабетической нефропатией, неизвестно, действует ли этот механизм, преодолевается или изменяется другими.
NGAL также участвует в развитии почек, индуцируя дифференцировку почечных предшественников в метанефрической мезенхиме в почечный эпителий [79]. NGAL может продуцироваться во многих органах, включая почки, печень, сердце, кишечник и различные популяции иммунных клеток, таких как Mφ или дендритные клетки [80,81], а также может быть одним из ключевых потенциальных модуляторов фенотипа макрофагов.
Было показано, что системная доставка NGAL оказывает защитное действие при ишемической ОПП за счет ингибирования гибели канальцевых клеток и индукции антиоксидантных генов [82]. NGAL играет решающую роль в пролиферации эпителия, ослабляя активацию PPARγ и в индукции экспрессии эпителиальных маркеров посредством активации мегалина и последующей активации сигнального пути PI3K / Akt [83].
Кроме того, сверхэкспрессия Mφ NGAL может индуцировать внутреннюю устойчивость к ишемии, вызывая защиту от ишемии-реперфузионного повреждения почек [84].Более того, Mφ, сверхэкспрессирующий IL-10, может защищать почки от ишемической AKI и улучшать восстановление тканей за счет индукции NGAL [85]. Подобно этому исследованию, было обнаружено, что у мышей NGAL KO было более тяжелое повреждение почек после ИР-индуцированной ОПП [86]. Более того, терапия NGAL эффективна при DN [87]. В этом исследовании терапия макрофагами NGAL клетками увеличивала противовоспалительную молекулу IL-10 и уменьшала провоспалительную молекулу TNF-α, что указывает на то, что терапия сверхэкспрессирующими NGAL макрофагами может уменьшить воспаление в DN.Этот противовоспалительный эффект может иметь как прямое терапевтическое действие на DN, так и в то же время способствовать стабилизации фенотипа M2.
Инфламмасома NLRP3 является наиболее изученной среди инфламмасом, мультибелковых цитозольных комплексов, которые составляют часть семейства NLR PRR [88,89]. NLRP3 участвует в патогенезе хронической болезни почек [90,91]. В ткани из биопсии почек человека повышенная экспрессия мРНК NLRP3 была обнаружена при диабетических заболеваниях почек. Кроме того, NLRP3 был связан с функцией почек, а ингибирование активации инфламмасом NLRP3 предотвращает воспаление и фиброз почек, по крайней мере частично, посредством подавления окислительного стресса при диабетической нефропатии [92].
Активация инфламмасомы NLRP3 играет критическую роль в поляризации макрофага M1 [93,94]. Активация NF-κB является отличительной чертой M1, и было показано, что она способствует повреждению почек при серповидном гломерулонефрите [95]. Кроме того, если активация NF-κB ингибируется в Mφ, они приобретают противовоспалительный фенотип и подавляют повреждение почек при его переносе в модели нефротоксического нефрита на крысах [96]. Напротив, NLRP3 также участвует в поляризации M2, поскольку NLRP3 трансактивирует промотор IL-4 для увеличения экспрессии IL-4 (характерной для фенотипа M2) в Mφ, происходящем из моноцитов периферической крови [97]. Кроме того, у мышей UUO было обнаружено, что инфламмасома NLRP3 активирует сигнальные пути NF-κB и IL-4 / STAT6 и способствует инфильтрации макрофагов M1 и M2 и повреждению почек [98]. Следовательно, это указывает на то, что нацеливание на инфламмасому NLRP3 при диабете может действовать на Mφ при диабетической нефропатии.
Кроме того, у мышей UUO было обнаружено, что инфламмасома NLRP3 активирует сигнальные пути NF-κB и IL-4 / STAT6 и способствует инфильтрации макрофагов M1 и M2 и повреждению почек [98]. Следовательно, это указывает на то, что нацеливание на инфламмасому NLRP3 при диабете может действовать на Mφ при диабетической нефропатии.
6. Выводы
Хотя роль различных фенотипов Mφ в фиброзе и эффективность терапии Mφ со стабильным фенотипом M2 (генетически модифицированным) были изучены, остается неясным, как фенотип M2 может стать профиброзным и каково влияние окружающей среды.
Факторы, которые модулируют S1P, NGAL или NLPR3, могут быть эффективны в контексте диабетической нефропатии из-за их способности изменять фенотип Mφ или соотношение между прорезолюцией и воспалительным Mφ.
Выражение признательности
Это исследование было поддержано грантами от Ministerio de Economiía y Competitividad, ссылка SAF2015–67770-R, присужденных G.H.
Вклад авторов
G.H. исследовал данные, внес свой вклад в заключение и написал рукопись.ПК. исследовал данные и написал рукопись. G.H. является гарантом этой работы и берет на себя ответственность за ее достоверность. Все авторы прочитали и согласились с опубликованной версией рукописи.
Финансирование
Это исследование не получало внешнего финансирования.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Ссылки
1. Коллинз А.Дж., Фоли Р.Н., Чаверс Б., Гилбертсон Д., Херцог К., Йохансен К., Kasiske B., Kutner N., Liu J., St. Peter W. и др. Система данных почек США Годовой отчет за 2011 год: Атлас хронической болезни почек и терминальной стадии почечной недостаточности в Соединенных Штатах. Являюсь. J. Kidney Dis. 2012; 59: e1 – e420. [PubMed] [Google Scholar] 2. Нгуен Д., Пинг Ф., Му В., Хилл П., Аткинс Р.С., Чадбан С.Дж. Накопление макрофагов при прогрессирующей диабетической нефропатии человека. Нефрология. 2006; 11: 226–231. DOI: 10.1111 / j.1440-1797.2006.00576.x. [PubMed] [CrossRef] [Google Scholar] 3. Клессенс К.Q.F., Zandbergen M., Wolterbeek R., Bruijn J.A., Rabelink T.J., Bajema I.M., IJpelaar D.H.T. Макрофаги при диабетической нефропатии у больных сахарным диабетом 2 типа. Нефрол. Набирать номер. Пересадка. 2017; 32: 1322–1329. DOI: 10.1093 / ndt / gfw260. [PubMed] [CrossRef] [Google Scholar] 4. Фурута Т., Сайто Т., Отака Т., Сома Дж., Обара К., Абэ К., Йошинага К. Роль макрофагов в диабетическом гломерулосклерозе. Являюсь. J. Kidney Dis. 1993; 21: 480–485. DOI: 10.1016 / S0272-6386 (12) 80393-3. [PubMed] [CrossRef] [Google Scholar] 5.Хирата К., Шиката К., Мацуда М., Акияма К., Сугимото Х., Кусиро М., Макино Х. Повышенная экспрессия селектинов в почках пациентов с диабетической нефропатией. Диабетология. 1998. 41: 185–192. DOI: 10.1007 / s001250050888. [PubMed] [CrossRef] [Google Scholar] 6. Боле А., Верманн М., Богеншютц О., Батц К., Мюллер К.А., Мюллер Г.А. Патогенез хронической почечной недостаточности при диабетической нефропатии. Расследование 488 случаев диабетического гломерулосклероза. Патол. Res. Практик. 1991; 187: 251–259.DOI: 10.1016 / S0344-0338 (11) 80780-6. [PubMed] [CrossRef] [Google Scholar] 7. Nguyen TQ, Tarnow L., Andersen S., Hovind P., Parving H.-H., Goldschmeding R., van Nieuwenhoven FA Экскреция фактора роста соединительной ткани мочевыводящих путей коррелирует с клиническими маркерами почечной недостаточности в большой популяции пациентов с диабетом 1 типа Пациенты с диабетической нефропатией. Уход за диабетом. 2006; 29: 83–88. DOI: 10.2337 / diacare.29.01.06.dc05-1670. [PubMed] [CrossRef] [Google Scholar] 8. Йонемото С., Мачигучи Т., Номура К., Минаката Т., Нанно М., Йошида Х. Корреляция тканевых макрофагов и экспрессии цитоскелетных белков с фиброзом почек у пациентов с сахарным диабетом. Clin. Exp. Нефрол. 2006; 10: 186–192. DOI: 10.1007 / s10157-006-0426-7. [PubMed] [CrossRef] [Google Scholar] 9. Каммингс Б.С., МакХоват Дж., Шнельманн Р.Г. Роль эндоплазматического ретикулума Ca 2 + -независимая фосфолипаза A2 в цисплатин-индуцированном апоптозе почечных клеток.
DOI: 10.1111 / j.1440-1797.2006.00576.x. [PubMed] [CrossRef] [Google Scholar] 3. Клессенс К.Q.F., Zandbergen M., Wolterbeek R., Bruijn J.A., Rabelink T.J., Bajema I.M., IJpelaar D.H.T. Макрофаги при диабетической нефропатии у больных сахарным диабетом 2 типа. Нефрол. Набирать номер. Пересадка. 2017; 32: 1322–1329. DOI: 10.1093 / ndt / gfw260. [PubMed] [CrossRef] [Google Scholar] 4. Фурута Т., Сайто Т., Отака Т., Сома Дж., Обара К., Абэ К., Йошинага К. Роль макрофагов в диабетическом гломерулосклерозе. Являюсь. J. Kidney Dis. 1993; 21: 480–485. DOI: 10.1016 / S0272-6386 (12) 80393-3. [PubMed] [CrossRef] [Google Scholar] 5.Хирата К., Шиката К., Мацуда М., Акияма К., Сугимото Х., Кусиро М., Макино Х. Повышенная экспрессия селектинов в почках пациентов с диабетической нефропатией. Диабетология. 1998. 41: 185–192. DOI: 10.1007 / s001250050888. [PubMed] [CrossRef] [Google Scholar] 6. Боле А., Верманн М., Богеншютц О., Батц К., Мюллер К.А., Мюллер Г.А. Патогенез хронической почечной недостаточности при диабетической нефропатии. Расследование 488 случаев диабетического гломерулосклероза. Патол. Res. Практик. 1991; 187: 251–259.DOI: 10.1016 / S0344-0338 (11) 80780-6. [PubMed] [CrossRef] [Google Scholar] 7. Nguyen TQ, Tarnow L., Andersen S., Hovind P., Parving H.-H., Goldschmeding R., van Nieuwenhoven FA Экскреция фактора роста соединительной ткани мочевыводящих путей коррелирует с клиническими маркерами почечной недостаточности в большой популяции пациентов с диабетом 1 типа Пациенты с диабетической нефропатией. Уход за диабетом. 2006; 29: 83–88. DOI: 10.2337 / diacare.29.01.06.dc05-1670. [PubMed] [CrossRef] [Google Scholar] 8. Йонемото С., Мачигучи Т., Номура К., Минаката Т., Нанно М., Йошида Х. Корреляция тканевых макрофагов и экспрессии цитоскелетных белков с фиброзом почек у пациентов с сахарным диабетом. Clin. Exp. Нефрол. 2006; 10: 186–192. DOI: 10.1007 / s10157-006-0426-7. [PubMed] [CrossRef] [Google Scholar] 9. Каммингс Б.С., МакХоват Дж., Шнельманн Р.Г. Роль эндоплазматического ретикулума Ca 2 + -независимая фосфолипаза A2 в цисплатин-индуцированном апоптозе почечных клеток. J. Pharmacol. Exp. Ther. 2004; 308: 921–928. DOI: 10.1124 / jpet.103.060541. [PubMed] [CrossRef] [Google Scholar] 10. Чоудхури П., Сакс С.Х., Ширин Н.С. Toll-подобные рецепторы TLR2 и TLR4 инициируют врожденный иммунный ответ эпителия почечных канальцев на бактериальные продукты. Clin. Exp. Иммунол. 2006. 145: 346–356. DOI: 10.1111 / j.1365-2249.2006.03116.x. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 11. Авад А.С., Кинси Г.Р., Хуцишвили К., Гао Т., Болтон В.К., Окуса М.Д. Хемокиновый рецептор моноцитов / макрофагов CCR2 опосредует диабетическое повреждение почек.Являюсь. J. Physiol. Рен. Physiol. 2011; 301: F1358 – F1366. DOI: 10.1152 / ajprenal.00332.2011. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 12. Теш Г.Х. Роль макрофагов в осложнениях диабета 2 типа. Clin. Exp. Pharmacol. Physiol. 2007; 34: 1016–1019. DOI: 10.1111 / j.1440-1681.2007.04729.x. [PubMed] [CrossRef] [Google Scholar] 13. Хасегава Г., Накано К., Савада М., Уно К., Шибаяма Ю., Йенага К., Кондо М. Возможная роль фактора некроза опухоли и интерлейкина-1 в развитии диабетической нефропатии.Kidney Int. 1991; 40: 1007–1012. DOI: 10.1038 / ки.1991.308. [PubMed] [CrossRef] [Google Scholar] 14. Калантариния К., Авад А.С., Сираджи Х.М. Концентрация TNF-альфа в моче и почках увеличивается до повышения альбуминурии у крыс с диабетом. Kidney Int. 2003. 64: 1208–1213. DOI: 10.1046 / j.1523-1755.2003.00237.x. [PubMed] [CrossRef] [Google Scholar] 15. Авад А.С., Ю Х., Гао Т., Купер Т.К., Недоспасов С.А., Вашер Дж., Уилкинсон П.Ф., Фаррелл Ф.X., Брайан Ривз В. Макрофагальный фактор некроза опухоли α опосредует диабетическое повреждение почек.Kidney Int. 2015; 88: 722–733. DOI: 10.1038 / ki.2015.162. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 16. Наварро-Гонсалес Дж. Ф., Мора-Фернандес К., Мурос де Фуэнтес М., Чахин Дж., Мендес М. Л., Гальего Э., Мака М., Дель Кастильо Н., Риверо А., Гетино М. А. и др. Влияние пентоксифиллина на функцию почек и экскрецию альбумина с мочой у пациентов с диабетическим заболеванием почек: исследование PREDIAN.
J. Pharmacol. Exp. Ther. 2004; 308: 921–928. DOI: 10.1124 / jpet.103.060541. [PubMed] [CrossRef] [Google Scholar] 10. Чоудхури П., Сакс С.Х., Ширин Н.С. Toll-подобные рецепторы TLR2 и TLR4 инициируют врожденный иммунный ответ эпителия почечных канальцев на бактериальные продукты. Clin. Exp. Иммунол. 2006. 145: 346–356. DOI: 10.1111 / j.1365-2249.2006.03116.x. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 11. Авад А.С., Кинси Г.Р., Хуцишвили К., Гао Т., Болтон В.К., Окуса М.Д. Хемокиновый рецептор моноцитов / макрофагов CCR2 опосредует диабетическое повреждение почек.Являюсь. J. Physiol. Рен. Physiol. 2011; 301: F1358 – F1366. DOI: 10.1152 / ajprenal.00332.2011. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 12. Теш Г.Х. Роль макрофагов в осложнениях диабета 2 типа. Clin. Exp. Pharmacol. Physiol. 2007; 34: 1016–1019. DOI: 10.1111 / j.1440-1681.2007.04729.x. [PubMed] [CrossRef] [Google Scholar] 13. Хасегава Г., Накано К., Савада М., Уно К., Шибаяма Ю., Йенага К., Кондо М. Возможная роль фактора некроза опухоли и интерлейкина-1 в развитии диабетической нефропатии.Kidney Int. 1991; 40: 1007–1012. DOI: 10.1038 / ки.1991.308. [PubMed] [CrossRef] [Google Scholar] 14. Калантариния К., Авад А.С., Сираджи Х.М. Концентрация TNF-альфа в моче и почках увеличивается до повышения альбуминурии у крыс с диабетом. Kidney Int. 2003. 64: 1208–1213. DOI: 10.1046 / j.1523-1755.2003.00237.x. [PubMed] [CrossRef] [Google Scholar] 15. Авад А.С., Ю Х., Гао Т., Купер Т.К., Недоспасов С.А., Вашер Дж., Уилкинсон П.Ф., Фаррелл Ф.X., Брайан Ривз В. Макрофагальный фактор некроза опухоли α опосредует диабетическое повреждение почек.Kidney Int. 2015; 88: 722–733. DOI: 10.1038 / ki.2015.162. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 16. Наварро-Гонсалес Дж. Ф., Мора-Фернандес К., Мурос де Фуэнтес М., Чахин Дж., Мендес М. Л., Гальего Э., Мака М., Дель Кастильо Н., Риверо А., Гетино М. А. и др. Влияние пентоксифиллина на функцию почек и экскрецию альбумина с мочой у пациентов с диабетическим заболеванием почек: исследование PREDIAN. Варенье. Soc. Нефрол. 2015; 26: 220–229. DOI: 10.1681 / ASN.2014010012. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 17.Gohda T., Niewczas M.A., Ficociello L.H., Walker W.H., Skupien J., Rosetti F., Cullere X., Johnson A.C., Crabtree G., Smiles A.M. и др. Циркулирующие рецепторы TNF 1 и 2 предсказывают 3 стадию ХБП при диабете 1 типа. Варенье. Soc. Нефрол. 2012; 23: 516–524. DOI: 10.1681 / ASN.2011060628. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 18. Niewczas M.A., Gohda T., Skupien J., Smiles A.M., Walker W.H., Rosetti F., Cullere X., Eckfeldt J.H., Doria A., Mayadas T.N. и др. Циркулирующие рецепторы TNF 1 и 2 предсказывают ТПН при диабете 2 типа.Варенье. Soc. Нефрол. 2012; 23: 507–515. DOI: 10.1681 / ASN.2011060627. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 19. Цзэн Л.-Ф., Сяо Ю., Сун Л. Краткий обзор механизмов, связанных с почечным фиброзом при диабетической нефропатии. Adv. Exp. Med. Биол. 2019; 1165: 49–79. [PubMed] [Google Scholar] 20. Brosius F.C. Новое понимание механизмов фиброза и склероза при диабетической нефропатии. Rev. Endocr. Метаб. Disord. 2008. 9: 245–254. DOI: 10.1007 / s11154-008-9100-6. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 21.Усуи Х.К., Шиката К., Сасаки М., Окада С., Мацуда М., Шиката Ю., Огава Д., Кидо Ю., Нагасе Р., Йозай К. и др. Мыши с дефицитом рецептора-поглотителя макрофагов устойчивы к диабетической нефропатии за счет уменьшения микровоспаления. Сахарный диабет. 2007. 56: 363–372. DOI: 10.2337 / db06-0359. [PubMed] [CrossRef] [Google Scholar] 22. Дистлер Дж. Х. У., Дьерфи А. Х., Рамануджам М., Уитфилд М. Л., Кёнигсхоф М., Лафятис Р. Общие и отдельные механизмы фиброза. Nat. Rev. Rheumatol. 2019; 15: 705–730.DOI: 10.1038 / s41584-019-0322-7. [PubMed] [CrossRef] [Google Scholar] 23. Джуджай С., Боор П. Клеточные и молекулярные механизмы фиброза почек. Мол. Asp. Med. 2019; 65: 16–36. DOI: 10.1016 / j.mam.2018.06.002. [PubMed] [CrossRef] [Google Scholar] 24. Ginhoux F.
Варенье. Soc. Нефрол. 2015; 26: 220–229. DOI: 10.1681 / ASN.2014010012. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 17.Gohda T., Niewczas M.A., Ficociello L.H., Walker W.H., Skupien J., Rosetti F., Cullere X., Johnson A.C., Crabtree G., Smiles A.M. и др. Циркулирующие рецепторы TNF 1 и 2 предсказывают 3 стадию ХБП при диабете 1 типа. Варенье. Soc. Нефрол. 2012; 23: 516–524. DOI: 10.1681 / ASN.2011060628. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 18. Niewczas M.A., Gohda T., Skupien J., Smiles A.M., Walker W.H., Rosetti F., Cullere X., Eckfeldt J.H., Doria A., Mayadas T.N. и др. Циркулирующие рецепторы TNF 1 и 2 предсказывают ТПН при диабете 2 типа.Варенье. Soc. Нефрол. 2012; 23: 507–515. DOI: 10.1681 / ASN.2011060627. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 19. Цзэн Л.-Ф., Сяо Ю., Сун Л. Краткий обзор механизмов, связанных с почечным фиброзом при диабетической нефропатии. Adv. Exp. Med. Биол. 2019; 1165: 49–79. [PubMed] [Google Scholar] 20. Brosius F.C. Новое понимание механизмов фиброза и склероза при диабетической нефропатии. Rev. Endocr. Метаб. Disord. 2008. 9: 245–254. DOI: 10.1007 / s11154-008-9100-6. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 21.Усуи Х.К., Шиката К., Сасаки М., Окада С., Мацуда М., Шиката Ю., Огава Д., Кидо Ю., Нагасе Р., Йозай К. и др. Мыши с дефицитом рецептора-поглотителя макрофагов устойчивы к диабетической нефропатии за счет уменьшения микровоспаления. Сахарный диабет. 2007. 56: 363–372. DOI: 10.2337 / db06-0359. [PubMed] [CrossRef] [Google Scholar] 22. Дистлер Дж. Х. У., Дьерфи А. Х., Рамануджам М., Уитфилд М. Л., Кёнигсхоф М., Лафятис Р. Общие и отдельные механизмы фиброза. Nat. Rev. Rheumatol. 2019; 15: 705–730.DOI: 10.1038 / s41584-019-0322-7. [PubMed] [CrossRef] [Google Scholar] 23. Джуджай С., Боор П. Клеточные и молекулярные механизмы фиброза почек. Мол. Asp. Med. 2019; 65: 16–36. DOI: 10.1016 / j.mam.2018.06.002. [PubMed] [CrossRef] [Google Scholar] 24. Ginhoux F. , Guilliams M. Онтогенез и гомеостаз тканевых макрофагов. Иммунитет. 2016; 44: 439–449. DOI: 10.1016 / j.immuni.2016.02.024. [PubMed] [CrossRef] [Google Scholar] 25. Viehmann S.F., Böhner A.M.C., Kurts C., Brähler S. Многогранная роль почечной системы мононуклеарных фагоцитов.Клетка. Иммунол. 2018; 330: 97–104. DOI: 10.1016 / j.cellimm.2018.04.009. [PubMed] [CrossRef] [Google Scholar] 26. Цао К., Харрис Д.К.Х., Ван Ю. Макрофаги при повреждении почек, воспалении и фиброзе. Физиология. 2015; 30: 183–194. DOI: 10.1152 / Physiol.00046.2014. [PubMed] [CrossRef] [Google Scholar] 27. Ван Н., Лян Х., Зен К. Молекулярные механизмы, влияющие на баланс поляризации макрофагов M1-M2. Фронт. Иммунол. 2014; 5: 614. DOI: 10.3389 / fimmu.2014.00614. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 29.Пураник А.С., Лиф И.А., Дженсен М.А., Хедаят А.Ф., Саад А., Ким К.В., Саадалла А.М., Вуллард Дж. Р., Кашьяп С., Textor S.C. и др. Резидентные в почках макрофаги способствуют созданию проангиогенной среды в нормальной и хронической ишемической почке мыши. Sci. Отчет 2018; 8: 13948. DOI: 10.1038 / s41598-018-31887-4. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 30. Согава Ю., Нагасу Х., Ивасе С., Ихория К., Итано С., Утида А., Кидокоро К., Танигучи С., Такахаши М., Сато М. и др. Инфильтрация макрофагов M1, но не M2, нарушается после односторонней обструкции мочеточника у мышей с дефицитом Nrf2.Sci. Отчет 2017; 7: 8801. DOI: 10.1038 / s41598-017-08054-2. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 31. Jiang Y., Wang Y., Ma P., An D., Zhao J., Liang S., Qin H. Миелоид-специфическое нацеливание Notch улучшает фиброз почек у мышей за счет уменьшения инфильтрации и активации макрофагов, полученных из костного мозга. Белковая клетка. 2019; 10: 196–210. DOI: 10.1007 / s13238-018-0527-6. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 32. Чжан X., Ян Ю., Чжао Ю. Фенотип макрофагов и его связь с функцией почек при диабетической нефропатии человека.
, Guilliams M. Онтогенез и гомеостаз тканевых макрофагов. Иммунитет. 2016; 44: 439–449. DOI: 10.1016 / j.immuni.2016.02.024. [PubMed] [CrossRef] [Google Scholar] 25. Viehmann S.F., Böhner A.M.C., Kurts C., Brähler S. Многогранная роль почечной системы мононуклеарных фагоцитов.Клетка. Иммунол. 2018; 330: 97–104. DOI: 10.1016 / j.cellimm.2018.04.009. [PubMed] [CrossRef] [Google Scholar] 26. Цао К., Харрис Д.К.Х., Ван Ю. Макрофаги при повреждении почек, воспалении и фиброзе. Физиология. 2015; 30: 183–194. DOI: 10.1152 / Physiol.00046.2014. [PubMed] [CrossRef] [Google Scholar] 27. Ван Н., Лян Х., Зен К. Молекулярные механизмы, влияющие на баланс поляризации макрофагов M1-M2. Фронт. Иммунол. 2014; 5: 614. DOI: 10.3389 / fimmu.2014.00614. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 29.Пураник А.С., Лиф И.А., Дженсен М.А., Хедаят А.Ф., Саад А., Ким К.В., Саадалла А.М., Вуллард Дж. Р., Кашьяп С., Textor S.C. и др. Резидентные в почках макрофаги способствуют созданию проангиогенной среды в нормальной и хронической ишемической почке мыши. Sci. Отчет 2018; 8: 13948. DOI: 10.1038 / s41598-018-31887-4. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 30. Согава Ю., Нагасу Х., Ивасе С., Ихория К., Итано С., Утида А., Кидокоро К., Танигучи С., Такахаши М., Сато М. и др. Инфильтрация макрофагов M1, но не M2, нарушается после односторонней обструкции мочеточника у мышей с дефицитом Nrf2.Sci. Отчет 2017; 7: 8801. DOI: 10.1038 / s41598-017-08054-2. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 31. Jiang Y., Wang Y., Ma P., An D., Zhao J., Liang S., Qin H. Миелоид-специфическое нацеливание Notch улучшает фиброз почек у мышей за счет уменьшения инфильтрации и активации макрофагов, полученных из костного мозга. Белковая клетка. 2019; 10: 196–210. DOI: 10.1007 / s13238-018-0527-6. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 32. Чжан X., Ян Ю., Чжао Ю. Фенотип макрофагов и его связь с функцией почек при диабетической нефропатии человека. PLoS ONE. 2019; 14: e0221991. DOI: 10.1371 / journal.pone.0221991. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 33. Zhang C., Zhang Y., Zhang C., Liu Y., Liu Y., Xu G. Пиоглитазон увеличивает экспрессию VEGFR3 и способствует активации макрофагов M2 через рецептор γ Mol, активируемый пролифератором пероксисом. Med. Отчет 2019; 19: 2740–2748. DOI: 10.3892 / mmr.2019.9945. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 34. Хасегава С., Накано Т., Торису К., Цучимото А., Эригути М., Харуяма Н., Масутани К., Казухико Цуруя К., Китазоно Т. Фактор роста эндотелия сосудов-С улучшает почечный интерстициальный фиброз посредством лимфангиогенеза при односторонней обструкции мочеточника у мышей. Лаборатория. Расследование. 2017; 97: 1439–1452. DOI: 10.1038 / labinvest.2017.77. [PubMed] [CrossRef] [Google Scholar] 35. Garibaldi S., Barisione C., Marengo B., Ameri P., Brunelli C., Balbi M., Ghigliotti G. Альбумин, модифицированный продуктами белков улучшенного окисления, индуцирует дифференцировку макрофагов RAW264.7 в дендритные клетки, которые модулируются тиолы клеточной поверхности.Токсины. 2017; 9: 27. DOI: 10.3390 / toxins
PLoS ONE. 2019; 14: e0221991. DOI: 10.1371 / journal.pone.0221991. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 33. Zhang C., Zhang Y., Zhang C., Liu Y., Liu Y., Xu G. Пиоглитазон увеличивает экспрессию VEGFR3 и способствует активации макрофагов M2 через рецептор γ Mol, активируемый пролифератором пероксисом. Med. Отчет 2019; 19: 2740–2748. DOI: 10.3892 / mmr.2019.9945. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 34. Хасегава С., Накано Т., Торису К., Цучимото А., Эригути М., Харуяма Н., Масутани К., Казухико Цуруя К., Китазоно Т. Фактор роста эндотелия сосудов-С улучшает почечный интерстициальный фиброз посредством лимфангиогенеза при односторонней обструкции мочеточника у мышей. Лаборатория. Расследование. 2017; 97: 1439–1452. DOI: 10.1038 / labinvest.2017.77. [PubMed] [CrossRef] [Google Scholar] 35. Garibaldi S., Barisione C., Marengo B., Ameri P., Brunelli C., Balbi M., Ghigliotti G. Альбумин, модифицированный продуктами белков улучшенного окисления, индуцирует дифференцировку макрофагов RAW264.7 в дендритные клетки, которые модулируются тиолы клеточной поверхности.Токсины. 2017; 9: 27. DOI: 10.3390 / toxins
27. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 36. Falke L.L., Gholizadeh S., Goldschmeding R., Kok R.J., Nguyen T.Q. Различное происхождение последствий миофибробластов для фиброза почек. Nat. Преподобный Нефрол. 2015; 11: 233–244. DOI: 10.1038 / nrneph.2014.246. [PubMed] [CrossRef] [Google Scholar] 37. Леблеу В.С., Тадури Г., О’Коннелл Дж., Тенг Ю., Кук В.Г., Вода С., Сугимото Х., Каллури Р. Происхождение и функция миофибробластов при фиброзе почек.Nat. Med. 2013; 19: 1047–1053. DOI: 10,1038 / нм 3218. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 38. Эдди А.А. Происхождение рубцовых миофибробластов почек. Nat. Med. 2013; 19: 964–966. DOI: 10,1038 / нм 3299. [PubMed] [CrossRef] [Google Scholar] 39. Ming-Kuen Tang P., Zhou S., Li C.-J., Liao J., Xiao J., Wang Q.-M., Lian G.-Y., Li J., Huang X. -R. , К.-Ф. и др. Протоонкогеновая тирозиновая протеинкиназа Src необходима для перехода от макрофагов к миофибробластам во время рубцевания почек. Kidney Int.2018 doi: 10.1016 / j.kint.2017.07.026. [PubMed] [CrossRef] [Google Scholar] 40. Ван Ю.Я., Цзян Х., Пань Дж., Хуан Х.Р., Ван Ю.С., Хуанг Х.Ф., То К.Ф., Николич-Патерсон Д.Дж., Лан Х.Й., Чен Дж. Х. Переход макрофагов в миофибробласты способствует интерстициальному фиброзу при хроническом повреждении почечного аллотрансплантата. Варенье. Soc. Нефрол. 2017; 28: 2053–2067. DOI: 10.1681 / ASN.2016050573. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 41. Мэн X.M., Ван С., Хуан X.R., Ян К., Сяо Дж., Чжан Ю., То К.Ф., Николич-Патерсон Д.Дж., Лан Х.Ю. Воспалительные макрофаги могут трансдифференцироваться в миофибробласты во время почечного фиброза. Cell Death Dis. 2016; 7: e2495. DOI: 10.1038 / cddis.2016.402. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 42. Ян Й., Фэн Х., Лю Х., Ван Й., Ху М., Цао К., Чжан З., Чжао Л., Чжан Дж., Го Р. и др. Изменение судьбы макрофагов, происходящих из костного мозга, улучшает фиброз почек на мышиной модели односторонней обструкции мочеточника. Нефрол. Набирать номер. Пересадка. 2019; 34: 1657–1668.DOI: 10.1093 / ndt / gfy381. [PubMed] [CrossRef] [Google Scholar] 43. Чоу Ф., Озолс Э., Николич-Патерсон Д.Дж., Аткинс Р.С., Теш Г.Х. Макрофаги при диабетической нефропатии 2 типа у мышей: корреляция с диабетическим состоянием и прогрессирующим повреждением почек. Kidney Int. 2004. 65: 116–128. DOI: 10.1111 / j.1523-1755.2004.00367.x. [PubMed] [CrossRef] [Google Scholar] 44. Окада С., Шиката К., Мацуда М., Огава Д., Усуи Х., Кидо Ю., Нагасе Р., Вада Дж., Шиката Ю., Макино Х. Мыши с дефицитом молекулы-1 межклеточной адгезии устойчивы к повреждение почек после индукции диабета.Сахарный диабет. 2003. 52: 2586–2593. DOI: 10,2337 / диабет. 52.10.2586. [PubMed] [CrossRef] [Google Scholar] 45. Ю Х., Гао Т., Купер Т.К., Брайан Ривз В., Авад А.С. Макрофаги непосредственно опосредуют диабетическое повреждение почек.
-R. , К.-Ф. и др. Протоонкогеновая тирозиновая протеинкиназа Src необходима для перехода от макрофагов к миофибробластам во время рубцевания почек. Kidney Int.2018 doi: 10.1016 / j.kint.2017.07.026. [PubMed] [CrossRef] [Google Scholar] 40. Ван Ю.Я., Цзян Х., Пань Дж., Хуан Х.Р., Ван Ю.С., Хуанг Х.Ф., То К.Ф., Николич-Патерсон Д.Дж., Лан Х.Й., Чен Дж. Х. Переход макрофагов в миофибробласты способствует интерстициальному фиброзу при хроническом повреждении почечного аллотрансплантата. Варенье. Soc. Нефрол. 2017; 28: 2053–2067. DOI: 10.1681 / ASN.2016050573. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 41. Мэн X.M., Ван С., Хуан X.R., Ян К., Сяо Дж., Чжан Ю., То К.Ф., Николич-Патерсон Д.Дж., Лан Х.Ю. Воспалительные макрофаги могут трансдифференцироваться в миофибробласты во время почечного фиброза. Cell Death Dis. 2016; 7: e2495. DOI: 10.1038 / cddis.2016.402. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 42. Ян Й., Фэн Х., Лю Х., Ван Й., Ху М., Цао К., Чжан З., Чжао Л., Чжан Дж., Го Р. и др. Изменение судьбы макрофагов, происходящих из костного мозга, улучшает фиброз почек на мышиной модели односторонней обструкции мочеточника. Нефрол. Набирать номер. Пересадка. 2019; 34: 1657–1668.DOI: 10.1093 / ndt / gfy381. [PubMed] [CrossRef] [Google Scholar] 43. Чоу Ф., Озолс Э., Николич-Патерсон Д.Дж., Аткинс Р.С., Теш Г.Х. Макрофаги при диабетической нефропатии 2 типа у мышей: корреляция с диабетическим состоянием и прогрессирующим повреждением почек. Kidney Int. 2004. 65: 116–128. DOI: 10.1111 / j.1523-1755.2004.00367.x. [PubMed] [CrossRef] [Google Scholar] 44. Окада С., Шиката К., Мацуда М., Огава Д., Усуи Х., Кидо Ю., Нагасе Р., Вада Дж., Шиката Ю., Макино Х. Мыши с дефицитом молекулы-1 межклеточной адгезии устойчивы к повреждение почек после индукции диабета.Сахарный диабет. 2003. 52: 2586–2593. DOI: 10,2337 / диабет. 52.10.2586. [PubMed] [CrossRef] [Google Scholar] 45. Ю Х., Гао Т., Купер Т.К., Брайан Ривз В., Авад А.С. Макрофаги непосредственно опосредуют диабетическое повреждение почек. Являюсь. J. Physiol. Рен. Physiol. 2013; 305: F1719 – F1727. DOI: 10.1152 / ajprenal.00141.2013. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 46. Чоу Ф.Ю., Николич-Патерсон Д.Д., Озолс Э., Аткинс Р.С., Теш Г.Х. Дефицит молекулы-1 межклеточной адгезии защищает от нефропатии у мышей db / db с диабетом 2 типа.Варенье. Soc. Нефрол. 2005; 16: 1711–1722. DOI: 10.1681 / ASN.2004070612. [PubMed] [CrossRef] [Google Scholar] 47. Чоу Ф.Ю., Николич-Патерсон Д.Дж., Ма Ф.Ю., Озолс Э., Роллинз Б.Дж., Теш Г.Х. Воспаление ткани, индуцированное хемоаттрактантным белком-1 моноцитов, имеет решающее значение для развития почечного повреждения, но не диабета 2 типа у тучных мышей db / db. Диабетология. 2007; 50: 471–480. DOI: 10.1007 / s00125-006-0497-8. [PubMed] [CrossRef] [Google Scholar] 48. Халлер Х., Бертрам А., Надровиц Ф., Менне Дж. Хемоаттрактантный белок-1 моноцитов и почки.Curr. Opin. Нефрол. Гипертоническая болезнь. 2016; 25: 42–49. DOI: 10.1097 / MNH.0000000000000186. [PubMed] [CrossRef] [Google Scholar] 49. Чоу Ф.Ю., Николич-Патерсон Д.Д., Озолс Э., Аткинс Р.С., Роллин Б.Дж., Теш Г.Х. Хемоаттрактантный белок-1 моноцитов способствует развитию диабетического поражения почек у мышей, получавших стрептозотоцин. Kidney Int. 2006; 69: 73–80. DOI: 10.1038 / sj.ki.5000014. [PubMed] [CrossRef] [Google Scholar] 50. Таширо К., Коянаги И., Сайто А., Симидзу А., Шике Т., Исигуро К., Коидзуми М., Фунабики К., Хорикоши С., Сирато И. и др. Уровни в моче моноцитарного хемоаттрактантного белка-1 (MCP-1) и интерлейкина-8 (IL-8) и почечные повреждения у пациентов с диабетической нефропатией 2 типа. J. Clin. Лаборатория. Анальный. 2002; 16: 1–4. DOI: 10.1002 / jcla.2057. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 51. Мацукава А., Хогабоам К.М., Лукач Н.В., Линкольн П.М., Стритер Р.М., Кункель С.Л. Эндогенный моноцитарный хемоаттрактантный белок-1 (MCP-1) защищает мышей в модели острого септического перитонита: перекрестное взаимодействие между MCP-1 и лейкотриеном B4.
Являюсь. J. Physiol. Рен. Physiol. 2013; 305: F1719 – F1727. DOI: 10.1152 / ajprenal.00141.2013. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 46. Чоу Ф.Ю., Николич-Патерсон Д.Д., Озолс Э., Аткинс Р.С., Теш Г.Х. Дефицит молекулы-1 межклеточной адгезии защищает от нефропатии у мышей db / db с диабетом 2 типа.Варенье. Soc. Нефрол. 2005; 16: 1711–1722. DOI: 10.1681 / ASN.2004070612. [PubMed] [CrossRef] [Google Scholar] 47. Чоу Ф.Ю., Николич-Патерсон Д.Дж., Ма Ф.Ю., Озолс Э., Роллинз Б.Дж., Теш Г.Х. Воспаление ткани, индуцированное хемоаттрактантным белком-1 моноцитов, имеет решающее значение для развития почечного повреждения, но не диабета 2 типа у тучных мышей db / db. Диабетология. 2007; 50: 471–480. DOI: 10.1007 / s00125-006-0497-8. [PubMed] [CrossRef] [Google Scholar] 48. Халлер Х., Бертрам А., Надровиц Ф., Менне Дж. Хемоаттрактантный белок-1 моноцитов и почки.Curr. Opin. Нефрол. Гипертоническая болезнь. 2016; 25: 42–49. DOI: 10.1097 / MNH.0000000000000186. [PubMed] [CrossRef] [Google Scholar] 49. Чоу Ф.Ю., Николич-Патерсон Д.Д., Озолс Э., Аткинс Р.С., Роллин Б.Дж., Теш Г.Х. Хемоаттрактантный белок-1 моноцитов способствует развитию диабетического поражения почек у мышей, получавших стрептозотоцин. Kidney Int. 2006; 69: 73–80. DOI: 10.1038 / sj.ki.5000014. [PubMed] [CrossRef] [Google Scholar] 50. Таширо К., Коянаги И., Сайто А., Симидзу А., Шике Т., Исигуро К., Коидзуми М., Фунабики К., Хорикоши С., Сирато И. и др. Уровни в моче моноцитарного хемоаттрактантного белка-1 (MCP-1) и интерлейкина-8 (IL-8) и почечные повреждения у пациентов с диабетической нефропатией 2 типа. J. Clin. Лаборатория. Анальный. 2002; 16: 1–4. DOI: 10.1002 / jcla.2057. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 51. Мацукава А., Хогабоам К.М., Лукач Н.В., Линкольн П.М., Стритер Р.М., Кункель С.Л. Эндогенный моноцитарный хемоаттрактантный белок-1 (MCP-1) защищает мышей в модели острого септического перитонита: перекрестное взаимодействие между MCP-1 и лейкотриеном B4. J. Immunol. 1999. 163: 6148–6154. [PubMed] [Google Scholar] 52. Мензис Р.И., Бут Дж. У. Р., Маллинз Дж. Дж., Бейли М. А., Тэм Ф. У. К., Норман Дж. Т., Анвин Р. Дж. Вызванная гипергликемией активация почечного рецептора P2X7 усиливает травмы, связанные с диабетом. EBioMedicine. 2017; 19: 73–83. DOI: 10.1016 / j.ebiom.2017.04.011. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 53. Menne J., Eulberg D., Beyer D., Baumann M., Saudek F., Valkusz Z., Rcek AW, Haller H. Ингибирование мотива-лиганда 2 СС с помощью эмаптикап-пегола (NOX-E36) у пациентов с диабетом 2 типа с альбуминурия.Нефрол. Набирать номер. Пересадка. 2016; 32: 307–315. DOI: 10.1093 / ndt / gfv459. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 54. Салливан Т., Мяо З., Дайраги Д.Дж., Красински А., Ван Й., Чжао Б.Н., Баумгарт Т., Эртл Л.С., Пеннелл А., Зейтц Л. и др. Антагонист CCR2 CCX140-B оказывает благоприятное воздействие на почки и гликемию у мышей с трансгенным диабетом человека с ноккином CCR2. Являюсь. J. Physiol. Рен. Physiol. 2013; 305: F1288 – F1297. DOI: 10.1152 / ajprenal.00316.2013. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 55.Минтон К. Макрофаги: фактор транскрипции, который можно назвать своим собственным. Nat. Rev. Immunol. 2011; 11: 74. DOI: 10.1038 / NRI2934. [PubMed] [CrossRef] [Google Scholar] 56. Zhu Y., Ling Y., Wang X. Алантолактон смягчает повреждение почек, вызванное диабетом, путем ингибирования воспалительного ответа, опосредованного высоким содержанием глюкозы, и инфильтрации макрофагов. Иммунофармакол. Иммунотоксикол. 2020; 42: 84–92. DOI: 10.1080 / 08
J. Immunol. 1999. 163: 6148–6154. [PubMed] [Google Scholar] 52. Мензис Р.И., Бут Дж. У. Р., Маллинз Дж. Дж., Бейли М. А., Тэм Ф. У. К., Норман Дж. Т., Анвин Р. Дж. Вызванная гипергликемией активация почечного рецептора P2X7 усиливает травмы, связанные с диабетом. EBioMedicine. 2017; 19: 73–83. DOI: 10.1016 / j.ebiom.2017.04.011. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 53. Menne J., Eulberg D., Beyer D., Baumann M., Saudek F., Valkusz Z., Rcek AW, Haller H. Ингибирование мотива-лиганда 2 СС с помощью эмаптикап-пегола (NOX-E36) у пациентов с диабетом 2 типа с альбуминурия.Нефрол. Набирать номер. Пересадка. 2016; 32: 307–315. DOI: 10.1093 / ndt / gfv459. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 54. Салливан Т., Мяо З., Дайраги Д.Дж., Красински А., Ван Й., Чжао Б.Н., Баумгарт Т., Эртл Л.С., Пеннелл А., Зейтц Л. и др. Антагонист CCR2 CCX140-B оказывает благоприятное воздействие на почки и гликемию у мышей с трансгенным диабетом человека с ноккином CCR2. Являюсь. J. Physiol. Рен. Physiol. 2013; 305: F1288 – F1297. DOI: 10.1152 / ajprenal.00316.2013. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 55.Минтон К. Макрофаги: фактор транскрипции, который можно назвать своим собственным. Nat. Rev. Immunol. 2011; 11: 74. DOI: 10.1038 / NRI2934. [PubMed] [CrossRef] [Google Scholar] 56. Zhu Y., Ling Y., Wang X. Алантолактон смягчает повреждение почек, вызванное диабетом, путем ингибирования воспалительного ответа, опосредованного высоким содержанием глюкозы, и инфильтрации макрофагов. Иммунофармакол. Иммунотоксикол. 2020; 42: 84–92. DOI: 10.1080 / 08
3.2020.1725039. [PubMed] [CrossRef] [Google Scholar] 57. Ян С., Ма К., Ву Х., Чжан Х., Юань Ф., Ян Г., Ян К., Цзя Л., Лян З., Кан Л. Текторигенин ослабляет диабетическую нефропатию, улучшая дисфункцию эндотелия сосудов за счет активации пути AdipoR1 / 2. Pharmacol. Res. 2020; 153: 104678. DOI: 10.1016 / j.phrs.2020.104678. [PubMed] [CrossRef] [Google Scholar] 58. Sun H., Tian J., Xian W. , Xie T., Yang X. Пентраксин-3 ослабляет повреждение почек при диабетической нефропатии, способствуя дифференцировке макрофагов M2. Воспаление. 2015; 38: 1739–1747. DOI: 10.1007 / s10753-015-0151-z. [PubMed] [CrossRef] [Google Scholar] 59.Cucak H., Nielsen Fink L., Højgaard Pedersen M., Rosendahl A. Лечение эналаприлом увеличивает количество Т-клеток и способствует локальной поляризации по отношению к M1-подобным макрофагам при диабетической нефропатии. Int. Иммунофармакол. 2015; 25: 30–42. DOI: 10.1016 / j.intimp.2015.01.003. [PubMed] [CrossRef] [Google Scholar] 60. Такахаши К., Ким Р.Х., Пасич Л., Хе Л., Нагасака С., Катагири Д., Мэй Т., Шимизу А., Харрис Р.С., Мерно Р.Л. Агонистические моноклональные антитела к CD148 ослабляют диабетическую нефропатию у мышей.Являюсь. J. Physiol. Рен. Physiol. 2020; 318: F647 – F659. DOI: 10.1152 / ajprenal.00288.2019. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 63. Гао К., Шен В., Цинь В., Чжэн К., Чжан М., Цзэн С., Ван С., Ван Дж., Чжу X., Лю З. Лечение мышей с диабетом db / db триптолидом: A новая терапия диабетической нефропатии. Нефрол. Набирать номер. Пересадка. 2010. 25: 3539–3547. DOI: 10,1093 / ndt / gfq245. [PubMed] [CrossRef] [Google Scholar] 64. Мохамед Р., Джаякумар К., Чен Ф., Фултон Д., Степп Д., Гансевоорт Р.Т., Рамеш Г. Терапия низкими дозами ИЛ-17 предотвращает и обращает вспять диабетическую нефропатию, метаболический синдром и связанный с ним фиброз органов. Варенье. Soc. Нефрол. 2016; 27: 745–765. DOI: 10.1681 / ASN.2014111136. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 65. Ким С.М., Ли С.Х., Ли А., Ким Д.Дж., Ким Ю.Г., Ким С.Й., Чжон К.Х., Ли Т.В., Им К.Г., Лим С.Дж. и др. Нацеливание микофенолятмофетила на Т-хелпер 17 снижает прогрессирование диабетической нефропатии. Пер. Res. J. Lab. Clin. Med. 2015; 166: 375–383.DOI: 10.1016 / j.trsl.2015.04.013. [PubMed] [CrossRef] [Google Scholar] 66. Eller K., Kirsch A., Wolf AM, Sopper S., Tagwerker A., Stanzl U., Wolf D., Patsch W.
, Xie T., Yang X. Пентраксин-3 ослабляет повреждение почек при диабетической нефропатии, способствуя дифференцировке макрофагов M2. Воспаление. 2015; 38: 1739–1747. DOI: 10.1007 / s10753-015-0151-z. [PubMed] [CrossRef] [Google Scholar] 59.Cucak H., Nielsen Fink L., Højgaard Pedersen M., Rosendahl A. Лечение эналаприлом увеличивает количество Т-клеток и способствует локальной поляризации по отношению к M1-подобным макрофагам при диабетической нефропатии. Int. Иммунофармакол. 2015; 25: 30–42. DOI: 10.1016 / j.intimp.2015.01.003. [PubMed] [CrossRef] [Google Scholar] 60. Такахаши К., Ким Р.Х., Пасич Л., Хе Л., Нагасака С., Катагири Д., Мэй Т., Шимизу А., Харрис Р.С., Мерно Р.Л. Агонистические моноклональные антитела к CD148 ослабляют диабетическую нефропатию у мышей.Являюсь. J. Physiol. Рен. Physiol. 2020; 318: F647 – F659. DOI: 10.1152 / ajprenal.00288.2019. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 63. Гао К., Шен В., Цинь В., Чжэн К., Чжан М., Цзэн С., Ван С., Ван Дж., Чжу X., Лю З. Лечение мышей с диабетом db / db триптолидом: A новая терапия диабетической нефропатии. Нефрол. Набирать номер. Пересадка. 2010. 25: 3539–3547. DOI: 10,1093 / ndt / gfq245. [PubMed] [CrossRef] [Google Scholar] 64. Мохамед Р., Джаякумар К., Чен Ф., Фултон Д., Степп Д., Гансевоорт Р.Т., Рамеш Г. Терапия низкими дозами ИЛ-17 предотвращает и обращает вспять диабетическую нефропатию, метаболический синдром и связанный с ним фиброз органов. Варенье. Soc. Нефрол. 2016; 27: 745–765. DOI: 10.1681 / ASN.2014111136. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 65. Ким С.М., Ли С.Х., Ли А., Ким Д.Дж., Ким Ю.Г., Ким С.Й., Чжон К.Х., Ли Т.В., Им К.Г., Лим С.Дж. и др. Нацеливание микофенолятмофетила на Т-хелпер 17 снижает прогрессирование диабетической нефропатии. Пер. Res. J. Lab. Clin. Med. 2015; 166: 375–383.DOI: 10.1016 / j.trsl.2015.04.013. [PubMed] [CrossRef] [Google Scholar] 66. Eller K., Kirsch A., Wolf AM, Sopper S., Tagwerker A., Stanzl U., Wolf D., Patsch W. , Rosenkranz AR, Eller P. Возможная роль регуляторных Т-клеток в обращении инсулинорезистентности, связанной с ожирением и диабетическая нефропатия. Сахарный диабет. 2011; 60: 2954–2962. DOI: 10.2337 / db11-0358. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 67. Хофкенс В., Сторм Г., Берг В.В.Д., Пост П.В. Подавление активации макрофагов M1 в пользу дифференцировки M2 за счет липосомного нацеливания глюкокортикоидов на синовиальную оболочку во время экспериментального артрита.Аня. Реум. Дис. 2011; 70: 70. DOI: 10.1136 / ard.2010.148973.11. [CrossRef] [Google Scholar] 68. Гордон С., Тейлор П.Р. Гетерогенность моноцитов и макрофагов. Nat. Rev. Immunol. 2005; 5: 953–964. DOI: 10.1038 / NRI1733. [PubMed] [CrossRef] [Google Scholar] 69. Ван Х., Яо Б., Ван Й., Фан Х, Ван С., Ню А., Ян Х., Фого А., Чжан М.З., Харрис Р.С. Макрофаг Циклооксигеназа-2 защищает от развития диабетической нефропатии. Сахарный диабет. 2017; 66: 494–504. DOI: 10.2337 / db16-0773. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 70.Zheng D., Wang Y., Cao Q., Lee V.W.S., Zheng G., Sun Y., Tan T.K., Wang Y., Alexander S.I., Harris D.C.H. Перелитые макрофаги улучшают повреждение поджелудочной железы и почек при сахарном диабете. Нефрон Эксп. Нефрол. 2011; 118: e87 – e99. DOI: 10,1159 / 000321034. [PubMed] [CrossRef] [Google Scholar] 71. Zhang X.L., Guo Y.F., Song Z.X., Zhou M. Витамин D предотвращает повреждение подоцитов посредством регуляции фенотипа макрофагов M1 / M2 у крыс с диабетической нефропатией. Эндокринология. 2014; 155: 4939–4950. DOI: 10.1210 / en.2014-1020. [PubMed] [CrossRef] [Google Scholar] 72. Ji L., Chen Y., Wang H., Zhang W., He L., Wu J., Liu Y. Избыточная экспрессия sirt6 способствует трансформации макрофагов M2, облегчая повреждение почек при диабетической нефропатии. Int. J. Oncol. 2019; 55: 103–115. [Бесплатная статья PMC] [PubMed] [Google Scholar] 73. Ндисанг Дж. Ф., Джадхав А. Геминотерапия улучшает функцию почек у самцов крыс с индуцированным стрептозотоцином диабетом: роль оси гемоксигеназы / предсердного натрийуретического пептида / адипонектина.
, Rosenkranz AR, Eller P. Возможная роль регуляторных Т-клеток в обращении инсулинорезистентности, связанной с ожирением и диабетическая нефропатия. Сахарный диабет. 2011; 60: 2954–2962. DOI: 10.2337 / db11-0358. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 67. Хофкенс В., Сторм Г., Берг В.В.Д., Пост П.В. Подавление активации макрофагов M1 в пользу дифференцировки M2 за счет липосомного нацеливания глюкокортикоидов на синовиальную оболочку во время экспериментального артрита.Аня. Реум. Дис. 2011; 70: 70. DOI: 10.1136 / ard.2010.148973.11. [CrossRef] [Google Scholar] 68. Гордон С., Тейлор П.Р. Гетерогенность моноцитов и макрофагов. Nat. Rev. Immunol. 2005; 5: 953–964. DOI: 10.1038 / NRI1733. [PubMed] [CrossRef] [Google Scholar] 69. Ван Х., Яо Б., Ван Й., Фан Х, Ван С., Ню А., Ян Х., Фого А., Чжан М.З., Харрис Р.С. Макрофаг Циклооксигеназа-2 защищает от развития диабетической нефропатии. Сахарный диабет. 2017; 66: 494–504. DOI: 10.2337 / db16-0773. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 70.Zheng D., Wang Y., Cao Q., Lee V.W.S., Zheng G., Sun Y., Tan T.K., Wang Y., Alexander S.I., Harris D.C.H. Перелитые макрофаги улучшают повреждение поджелудочной железы и почек при сахарном диабете. Нефрон Эксп. Нефрол. 2011; 118: e87 – e99. DOI: 10,1159 / 000321034. [PubMed] [CrossRef] [Google Scholar] 71. Zhang X.L., Guo Y.F., Song Z.X., Zhou M. Витамин D предотвращает повреждение подоцитов посредством регуляции фенотипа макрофагов M1 / M2 у крыс с диабетической нефропатией. Эндокринология. 2014; 155: 4939–4950. DOI: 10.1210 / en.2014-1020. [PubMed] [CrossRef] [Google Scholar] 72. Ji L., Chen Y., Wang H., Zhang W., He L., Wu J., Liu Y. Избыточная экспрессия sirt6 способствует трансформации макрофагов M2, облегчая повреждение почек при диабетической нефропатии. Int. J. Oncol. 2019; 55: 103–115. [Бесплатная статья PMC] [PubMed] [Google Scholar] 73. Ндисанг Дж. Ф., Джадхав А. Геминотерапия улучшает функцию почек у самцов крыс с индуцированным стрептозотоцином диабетом: роль оси гемоксигеназы / предсердного натрийуретического пептида / адипонектина. Эндокринология. 2014; 155: 215–229.DOI: 10.1210 / en.2013-1050. [PubMed] [CrossRef] [Google Scholar] 74. Guiteras R., Sola A., Flaquer M., Hotter G., Torras J., Grinyó J.M., Cruzado J.M. Макрофаги, сверхэкспрессирующие NGAL, улучшили фиброз почек в модели мышей UUO. Клетка. Physiol. Биохим. 2017; 42: 1945–1960. DOI: 10,1159 / 000479835. [PubMed] [CrossRef] [Google Scholar] 75. Wang L., Xing X.P., Holmes A., Wadham C., Gamble J.R., Vadas M.A., Xia P. Активация пути передачи сигналов сфингозинкиназы высоким содержанием глюкозы опосредует провоспалительный фенотип эндотелиальных клеток.Circ. Res. 2005; 97: 891–899. DOI: 10.1161 / 01.RES.0000187469.82595.15. [PubMed] [CrossRef] [Google Scholar] 76. Ягобиан Д., Дон Энтони С., Ягобиан С., Чен X., Поллок С.А., Саад С. Повышенный уровень сфингозин-1-фосфата опосредует воспаление и фиброз при повреждении канальцев при диабетической нефропатии. Clin. Exp. Pharmacol. Physiol. 2016; 43: 56–66. DOI: 10.1111 / 1440-1681.12494. [PubMed] [CrossRef] [Google Scholar] 77. Дельбридж М.С., Шреста Б.М., Рафтери А.Т., Эль Нахас А.М., Хейлор Дж.Л.Снижение ишемического реперфузионного повреждения в почке крысы с помощью FTY720, синтетического производного сфингозина.Трансплантация. 2007. 84: 187–195. DOI: 10.1097 / 01.tp.0000269794.74990.da. [PubMed] [CrossRef] [Google Scholar] 78. Sola A., Weigert A., Jung M., Vinuesa E., Brecht K., Weis N., Brüne B., Borregaard N., Hotter G. Передача сигналов сфингозин-1-фосфата индуцирует продукцию Lcn-2 макрофагами. способствовать регенерации почек. J. Pathol. 2011; 225: 597–608. DOI: 10.1002 / путь.2982. [PubMed] [CrossRef] [Google Scholar] 79. Ян Дж., Гетц Д., Ли Дж. Ю., Ван В., Мори К., Сетлик Д., Ду Т., Эрдджумент-Бромаж Х., Темпст П., Стронг Р. и др. Путь доставки железа, опосредованный липокалином. Мол. Клетка. 2002; 10: 1045–1056. DOI: 10.1016 / S1097-2765 (02) 00710-4. [PubMed] [CrossRef] [Google Scholar] 80. Торсвик С., Бакке И., ван Билен Гранлунд А., Ройсет Э.С., Дамос Дж. К., Эствик А. Е., Сандвик А.
Эндокринология. 2014; 155: 215–229.DOI: 10.1210 / en.2013-1050. [PubMed] [CrossRef] [Google Scholar] 74. Guiteras R., Sola A., Flaquer M., Hotter G., Torras J., Grinyó J.M., Cruzado J.M. Макрофаги, сверхэкспрессирующие NGAL, улучшили фиброз почек в модели мышей UUO. Клетка. Physiol. Биохим. 2017; 42: 1945–1960. DOI: 10,1159 / 000479835. [PubMed] [CrossRef] [Google Scholar] 75. Wang L., Xing X.P., Holmes A., Wadham C., Gamble J.R., Vadas M.A., Xia P. Активация пути передачи сигналов сфингозинкиназы высоким содержанием глюкозы опосредует провоспалительный фенотип эндотелиальных клеток.Circ. Res. 2005; 97: 891–899. DOI: 10.1161 / 01.RES.0000187469.82595.15. [PubMed] [CrossRef] [Google Scholar] 76. Ягобиан Д., Дон Энтони С., Ягобиан С., Чен X., Поллок С.А., Саад С. Повышенный уровень сфингозин-1-фосфата опосредует воспаление и фиброз при повреждении канальцев при диабетической нефропатии. Clin. Exp. Pharmacol. Physiol. 2016; 43: 56–66. DOI: 10.1111 / 1440-1681.12494. [PubMed] [CrossRef] [Google Scholar] 77. Дельбридж М.С., Шреста Б.М., Рафтери А.Т., Эль Нахас А.М., Хейлор Дж.Л.Снижение ишемического реперфузионного повреждения в почке крысы с помощью FTY720, синтетического производного сфингозина.Трансплантация. 2007. 84: 187–195. DOI: 10.1097 / 01.tp.0000269794.74990.da. [PubMed] [CrossRef] [Google Scholar] 78. Sola A., Weigert A., Jung M., Vinuesa E., Brecht K., Weis N., Brüne B., Borregaard N., Hotter G. Передача сигналов сфингозин-1-фосфата индуцирует продукцию Lcn-2 макрофагами. способствовать регенерации почек. J. Pathol. 2011; 225: 597–608. DOI: 10.1002 / путь.2982. [PubMed] [CrossRef] [Google Scholar] 79. Ян Дж., Гетц Д., Ли Дж. Ю., Ван В., Мори К., Сетлик Д., Ду Т., Эрдджумент-Бромаж Х., Темпст П., Стронг Р. и др. Путь доставки железа, опосредованный липокалином. Мол. Клетка. 2002; 10: 1045–1056. DOI: 10.1016 / S1097-2765 (02) 00710-4. [PubMed] [CrossRef] [Google Scholar] 80. Торсвик С., Бакке И., ван Билен Гранлунд А., Ройсет Э.С., Дамос Дж. К., Эствик А. Е., Сандвик А. К. Экспрессия липокалина, связанного с желатиназой нейтрофилов (NGAL), в кишечнике при болезни Крона. Клетка. Tissue Res. 2018; 374: 339–348. DOI: 10.1007 / s00441-018-2860-8. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 81.Чакраборти С., Каур С., Гуха С., Батра С.К. Многогранная роль липокалина, связанного с желатиназой нейтрофилов (NGAL), в воспалении и раке. Биохим. Биофиз. Acta. 2012; 1826: 129–169. DOI: 10.1016 / j.bbcan.2012.03.008. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 82. Мори К., Ли Х.Т., Рапопорт Д., Дрекслер И.Р., Фостер К., Ян Дж., Шмидт-Отт К.М., Чен Х., Джау Ю.Л., Вайс С. и др. Эндоцитарная доставка комплекса липокалин-сидерофор-железо спасает почку от ишемического реперфузионного повреждения.J. Clin. Расследование. 2005. 115: 610–621. DOI: 10,1172 / JCI23056. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 83. Jung M., Brüne B., von Knethen A., Guiteras R., Cruzado J.M., Hotter G., Sola A. Липокалин-2 отменяет остановку клеточного цикла эпителия за счет ингибирования PPARγ. Лаборатория. Расследование. 2018; 98: 1408–1422. DOI: 10.1038 / s41374-018-0098-4. [PubMed] [CrossRef] [Google Scholar] 84. Юнг М., Брюне Б., Хоттер Г., Сола А. Липокалин-2, полученный из макрофагов, вносит свой вклад в механизмы ишемической устойчивости, защищая от повреждения почек.Sci. Отчет 2016; 6: 21950. DOI: 10,1038 / srep21950. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 85. Jung M., Sola A., Hughes J., Kluth D.C., Vinuesa E., Viñas J.L., Pérez-Ladaga A., Hotter G. Инфузия клеток, экспрессирующих IL-10, защищает от ишемии почек за счет индукции липокалина-2. Kidney Int. 2012; 81: 969–982. DOI: 10.1038 / ki.2011.446. [PubMed] [CrossRef] [Google Scholar] 86. Ли С.А., Ноэль С., Курцхаген Дж.Т., Садасивам М., Пьерорацио П.М., Аренд Л.Дж., Хамад А.Р., Рабб Х. CD4 + Т-клеточный NGAL изменяет исход ишемической острой почечной травмы.J. Immunol. 2020; 204: 586–595. DOI: 10.4049 / jimmunol.1
К. Экспрессия липокалина, связанного с желатиназой нейтрофилов (NGAL), в кишечнике при болезни Крона. Клетка. Tissue Res. 2018; 374: 339–348. DOI: 10.1007 / s00441-018-2860-8. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 81.Чакраборти С., Каур С., Гуха С., Батра С.К. Многогранная роль липокалина, связанного с желатиназой нейтрофилов (NGAL), в воспалении и раке. Биохим. Биофиз. Acta. 2012; 1826: 129–169. DOI: 10.1016 / j.bbcan.2012.03.008. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 82. Мори К., Ли Х.Т., Рапопорт Д., Дрекслер И.Р., Фостер К., Ян Дж., Шмидт-Отт К.М., Чен Х., Джау Ю.Л., Вайс С. и др. Эндоцитарная доставка комплекса липокалин-сидерофор-железо спасает почку от ишемического реперфузионного повреждения.J. Clin. Расследование. 2005. 115: 610–621. DOI: 10,1172 / JCI23056. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 83. Jung M., Brüne B., von Knethen A., Guiteras R., Cruzado J.M., Hotter G., Sola A. Липокалин-2 отменяет остановку клеточного цикла эпителия за счет ингибирования PPARγ. Лаборатория. Расследование. 2018; 98: 1408–1422. DOI: 10.1038 / s41374-018-0098-4. [PubMed] [CrossRef] [Google Scholar] 84. Юнг М., Брюне Б., Хоттер Г., Сола А. Липокалин-2, полученный из макрофагов, вносит свой вклад в механизмы ишемической устойчивости, защищая от повреждения почек.Sci. Отчет 2016; 6: 21950. DOI: 10,1038 / srep21950. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 85. Jung M., Sola A., Hughes J., Kluth D.C., Vinuesa E., Viñas J.L., Pérez-Ladaga A., Hotter G. Инфузия клеток, экспрессирующих IL-10, защищает от ишемии почек за счет индукции липокалина-2. Kidney Int. 2012; 81: 969–982. DOI: 10.1038 / ki.2011.446. [PubMed] [CrossRef] [Google Scholar] 86. Ли С.А., Ноэль С., Курцхаген Дж.Т., Садасивам М., Пьерорацио П.М., Аренд Л.Дж., Хамад А.Р., Рабб Х. CD4 + Т-клеточный NGAL изменяет исход ишемической острой почечной травмы.J. Immunol. 2020; 204: 586–595. DOI: 10.4049 / jimmunol.1
7. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 87. Гитерас Р., Сола А., Флакер М., Манонеллес А., Хоттер Г., Крузадо Дж. М. Изучение терапии макрофагальными клетками при диабетической болезни почек. J. Cell. Мол. Med. 2019; 23: 841–851. DOI: 10.1111 / jcmm.13983. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 88. Муссо Г., Де Мичели Ф., Бонджованни Д., Паренте Р., Фрамарин Л., Леоне Н., Беррутти М., Гамбино Р., Кассадер М., Кохни С. и др.Новые фармакологические агенты, направленные на воспаление и фиброз при заболевании почек, не связанном с алкогольным стеатогепатитом. Clin. Гастроэнтерол. Гепатол. 2017; 15: 972–985. DOI: 10.1016 / j.cgh.2016.08.002. [PubMed] [CrossRef] [Google Scholar] 89. Lusarczyk J., Trojan E., Głombik K., Piotrowska A., Budziszewska B., Kubera M., Popiołek-Barczyk K., Lasoń W., Mika J., Basta-Kaim A. Нацеливание на пути, связанные с инфламмасомой NLRP3 через подавленную лечением тианептином поляризацию микроглии до фенотипа M1 в культурах, стимулированных липополисахаридами.Int. J. Mol. Sci. 2018; 19: 1965. DOI: 10.3390 / ijms1
Гитерас Р., Сола А., Флакер М., Манонеллес А., Хоттер Г., Крузадо Дж. М. Изучение терапии макрофагальными клетками при диабетической болезни почек. J. Cell. Мол. Med. 2019; 23: 841–851. DOI: 10.1111 / jcmm.13983. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 88. Муссо Г., Де Мичели Ф., Бонджованни Д., Паренте Р., Фрамарин Л., Леоне Н., Беррутти М., Гамбино Р., Кассадер М., Кохни С. и др.Новые фармакологические агенты, направленные на воспаление и фиброз при заболевании почек, не связанном с алкогольным стеатогепатитом. Clin. Гастроэнтерол. Гепатол. 2017; 15: 972–985. DOI: 10.1016 / j.cgh.2016.08.002. [PubMed] [CrossRef] [Google Scholar] 89. Lusarczyk J., Trojan E., Głombik K., Piotrowska A., Budziszewska B., Kubera M., Popiołek-Barczyk K., Lasoń W., Mika J., Basta-Kaim A. Нацеливание на пути, связанные с инфламмасомой NLRP3 через подавленную лечением тианептином поляризацию микроглии до фенотипа M1 в культурах, стимулированных липополисахаридами.Int. J. Mol. Sci. 2018; 19: 1965. DOI: 10.3390 / ijms1
65. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 90. Вилайсане А., Чун Дж., Симон М.Э., Ван В., Чин Р., Хирота С., Ли Ю., Кларк С.А., Чопп Дж., Трпков К. и др. Инфламмасома NLRP3 способствует воспалению почек и способствует развитию ХБП. Варенье. Soc. Нефрол. 2010; 21: 1732–1744. DOI: 10.1681 / ASN.2010020143. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 91. Андерс Х.Дж., Суарес-Альварес Б., Григореску М., Форесто-Нето О., Steiger S., Desai J., Marschner J.A., Honarpisheh M., Shi C., Jordan J., et al. Фенотип макрофагов и компонент инфламмасомы NLRP3 вносят вклад в связанное с нефрокальцинозом хроническое заболевание почек независимо от IL-1-опосредованного повреждения ткани. Kidney Int. 2018; 93: 656–669. DOI: 10.1016 / j.kint.2017.09.022. [PubMed] [CrossRef] [Google Scholar] 93. Конос С.А., Чен К.В., Де Нардо Д., Хара Х., Уайтхед Л., Нуньес Г., Мастерс С.Л., Мерфи Дж. М., Шредер К., Во Д. Л. и др. Активный MLKL запускает инфламмасому NLRP3 клеточно-тринитарным образом. Proc. Natl. Акад. Sci. США. 2017; 114: E961 – E969. DOI: 10.1073 / pnas.1613305114. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 94. Zhang L., Fan Y., Su H., Wu L., Huang Y., Zhao L., Han B., Shu G., Xiang M., Yang JM. Метиловый эфир хлорогеновой кислоты оказывает сильное противовоспалительное действие за счет ингибирования путь COX-2 / NLRP3 / NF-kappaB. Food Funct. 2018; 9: 6155–6164. DOI: 10.1039 / C8FO01281D. [PubMed] [CrossRef] [Google Scholar] 95. Томита Н., Моришита Р., Лан Х.Ю., Ямамото К., Хашизуме М., Нотаке М., Тоёсава К., Фудзитани Б., Му В., Николич-Патерсон Д.Дж. и др. Введение in vivo приманки ядерного фактора транскрипции — каппаВ подавляет экспериментальный серповидный гломерулонефрит. Варенье. Soc. Нефрол. 2000; 11: 1244–1252. [PubMed] [Google Scholar] 96. Wilson H.M., Chettibi S., Jobin C., Walbaum D., Rees A.J., Kluth D.C. Ингибирование ядерного фактора макрофагов-κb приводит к доминантному противовоспалительному фенотипу, который ослабляет воспаление клубочков in vivo. Являюсь. J. Pathol. 2005. 167: 27–37. DOI: 10.1016 / S0002-9440 (10) 62950-1. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 97. Лю Ю., Гао X., Мяо Ю., Ван Ю., Ван Х., Ченг З., Ван X., Цзин X., Цзя Л., Дай Л. и др. NLRP3 регулирует поляризацию макрофага M2 посредством активации IL-4 при астме. Биохим. J. 2018; 475: 1995–2008. DOI: 10.1042 / BCJ20180086. [PubMed] [CrossRef] [Google Scholar] 98. Zhou Y., Zhu X., Wang X., Peng Y., Du J., Yin H., Yang H., Ni X., Zhang W. H 2 S облегчает повреждение почек и фиброз в ответ на одностороннее поражение мочеточника. обструкция путем регулирования инфильтрации макрофагов посредством ингибирования передачи сигналов NLRP3.Exp. Cell Res. 2020; 387: 111779. DOI: 10.1016 / j.yexcr.2019.111779. [PubMed] [CrossRef] [Google Scholar]
Proc. Natl. Акад. Sci. США. 2017; 114: E961 – E969. DOI: 10.1073 / pnas.1613305114. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 94. Zhang L., Fan Y., Su H., Wu L., Huang Y., Zhao L., Han B., Shu G., Xiang M., Yang JM. Метиловый эфир хлорогеновой кислоты оказывает сильное противовоспалительное действие за счет ингибирования путь COX-2 / NLRP3 / NF-kappaB. Food Funct. 2018; 9: 6155–6164. DOI: 10.1039 / C8FO01281D. [PubMed] [CrossRef] [Google Scholar] 95. Томита Н., Моришита Р., Лан Х.Ю., Ямамото К., Хашизуме М., Нотаке М., Тоёсава К., Фудзитани Б., Му В., Николич-Патерсон Д.Дж. и др. Введение in vivo приманки ядерного фактора транскрипции — каппаВ подавляет экспериментальный серповидный гломерулонефрит. Варенье. Soc. Нефрол. 2000; 11: 1244–1252. [PubMed] [Google Scholar] 96. Wilson H.M., Chettibi S., Jobin C., Walbaum D., Rees A.J., Kluth D.C. Ингибирование ядерного фактора макрофагов-κb приводит к доминантному противовоспалительному фенотипу, который ослабляет воспаление клубочков in vivo. Являюсь. J. Pathol. 2005. 167: 27–37. DOI: 10.1016 / S0002-9440 (10) 62950-1. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar] 97. Лю Ю., Гао X., Мяо Ю., Ван Ю., Ван Х., Ченг З., Ван X., Цзин X., Цзя Л., Дай Л. и др. NLRP3 регулирует поляризацию макрофага M2 посредством активации IL-4 при астме. Биохим. J. 2018; 475: 1995–2008. DOI: 10.1042 / BCJ20180086. [PubMed] [CrossRef] [Google Scholar] 98. Zhou Y., Zhu X., Wang X., Peng Y., Du J., Yin H., Yang H., Ni X., Zhang W. H 2 S облегчает повреждение почек и фиброз в ответ на одностороннее поражение мочеточника. обструкция путем регулирования инфильтрации макрофагов посредством ингибирования передачи сигналов NLRP3.Exp. Cell Res. 2020; 387: 111779. DOI: 10.1016 / j.yexcr.2019.111779. [PubMed] [CrossRef] [Google Scholar]
Аутофагия и метаболические изменения при хронической болезни почек, связанной с ожирением | Нефрологическая диализная трансплантация
Аннотация
Ожирение является долговременным источником клеточного стресса, предрасполагающего к хронической болезни почек (ХБП). Аутофагия — это гомеостатический механизм контроля качества клеток посредством утилизации и переработки клеточных компонентов.Во время клеточного стресса аутофагия предоставляет механизмы для управления стрессом, избирательно избавляя клетку от накопления потенциально токсичных белков, липидов и органелл. Используемые адаптивные процессы могут различаться для разных типов клеток и выборочно приспосабливаться к поражению, вызывая компоненты основного аппарата аутофагии, используемые клетками, не находясь под принуждением. В этом обзоре мы обсудим аутофагические реакции органов на клеточные стрессоры, такие как диета с высоким содержанием жиров, ожирение и диабет, и то, как эти механизмы могут предотвращать или способствовать прогрессированию заболевания.Выявление ранних клеточных механизмов возникновения почечных осложнений, связанных с ожирением и диабетом, может открыть путь для будущих терапевтических вмешательств.
Аутофагия — это гомеостатический механизм контроля качества клеток посредством утилизации и переработки клеточных компонентов.Во время клеточного стресса аутофагия предоставляет механизмы для управления стрессом, избирательно избавляя клетку от накопления потенциально токсичных белков, липидов и органелл. Используемые адаптивные процессы могут различаться для разных типов клеток и выборочно приспосабливаться к поражению, вызывая компоненты основного аппарата аутофагии, используемые клетками, не находясь под принуждением. В этом обзоре мы обсудим аутофагические реакции органов на клеточные стрессоры, такие как диета с высоким содержанием жиров, ожирение и диабет, и то, как эти механизмы могут предотвращать или способствовать прогрессированию заболевания.Выявление ранних клеточных механизмов возникновения почечных осложнений, связанных с ожирением и диабетом, может открыть путь для будущих терапевтических вмешательств.
ВВЕДЕНИЕ
Ожирение продолжает оставаться широко распространенной проблемой общественного здравоохранения. В США распространенность ожирения удвоилась с 1980 по 2002 год, затронув примерно каждого третьего взрослого [1]. Жировая ткань предназначена не только для хранения энергии, но и представляет собой сложную эндокринную железу, которая взаимодействует с другими органами.Во многом благодаря этим взаимодействиям ожирение увеличивает вероятность различных заболеваний и ассоциируется с диабетом, гипертонией, протеинурией и артросклерозом, факторами риска хронической болезни почек (ХБП). Несколько исследований подтверждают эту связь между ожирением и заболеванием почек [2–4], включая недавний метаанализ, который продемонстрировал вклад ожирения в прогрессирующую потерю функции почек у пациентов с ХБП [5]. В этом обзоре мы кратко опишем связь между ожирением и регуляцией аутофагии, а также роль, которую аутофагия играет при заболеваниях почек, связанных с ожирением и диабетом.
ОЖИРЕНИЕ
Крысы Zucker с генетическим ожирением демонстрируют ранние изменения гемодинамики почек, включая увеличение скорости клубочковой фильтрации (СКФ) и альбуминурию, наряду с изменениями морфологии почек, включая увеличение клубочков, расширение мезангиальных клеток и повреждение подоцитов. Эти гемодинамические изменения обратимы при ограничении калорийности [6], как и структурные изменения [7]. У пациентов повышенный индекс массы тела (ИМТ) коррелирует не только с экскрецией альбумина, но и с ограничением калорийности или бариатрической хирургией, поскольку средство снижения веса снижает протеинурию [8].Воздействие на почки, помимо гемодинамических, структурных и патологических изменений, заключается в том, что ожирение увеличивает риск ХБП. Другие сопутствующие заболевания включают метаболический синдром, высокое кровяное давление, сахарный диабет и сердечно-сосудистые заболевания [9].
Связующим звеном между избытком калорий и ХЗП является датчик питательных веществ, 5′-АМФ-активированная протеинкиназа (AMPK). AMPK активируется, когда соотношение AMP (ADP): ATP увеличивается, например, во время ограничения калорийности или физических упражнений. AMPK также реагирует на адипокин, адипонектин, который, как было показано, снижается у тучных людей с инсулинорезистентностью.Мыши с дефицитом адипонектина демонстрируют низкую активность AMPK, альбуминурию и стирание отростков стопы подоцитов. Индукция агонистами активности AMPK ослабляет эти патологические состояния [10]. Одним из центральных клеточных гомеостатических механизмов, реагирующих на AMPK, является аутофагия, и появляется все больше литературы, связывающей аутофагию с хронической дисфункцией органов.
ОБЩАЯ АВТОФАГИЯ
Аутофагия — это консервативный катаболический механизм, необходимый для клеточного гомеостатического контроля качества и регенерации, а также механизма клеточной реакции на стресс. Существует три типа аутофагии, которые различаются по механизму доставки груза к лизосомам: макроаутофагия, шаперон-опосредованная аутофагия (CMA) и микроаутофагия. Макроаутофагия требует образования везикулы с двойной мембраной, аутофагосомы, которая при образовании секвестрирует цитозольные компоненты для доставки в лизосомы (Рисунок 1). CMA представляет собой форму селективной аутофагии с использованием шаперона теплового шока 70 кДа, hsc70, который распознает мотив пентапептида KFERQ при неправильной укладке белковых комплексов.Этот комплекс hsc70-белок связывается с лизосомальным рецепторным комплексом, разворачивается и перемещается в лизосому [11–13]. При микроаутофагии цитозольный материал непосредственно поглощается инвагинацией лизосомальной мембраны [14].
Существует три типа аутофагии, которые различаются по механизму доставки груза к лизосомам: макроаутофагия, шаперон-опосредованная аутофагия (CMA) и микроаутофагия. Макроаутофагия требует образования везикулы с двойной мембраной, аутофагосомы, которая при образовании секвестрирует цитозольные компоненты для доставки в лизосомы (Рисунок 1). CMA представляет собой форму селективной аутофагии с использованием шаперона теплового шока 70 кДа, hsc70, который распознает мотив пентапептида KFERQ при неправильной укладке белковых комплексов.Этот комплекс hsc70-белок связывается с лизосомальным рецепторным комплексом, разворачивается и перемещается в лизосому [11–13]. При микроаутофагии цитозольный материал непосредственно поглощается инвагинацией лизосомальной мембраны [14].
РИСУНОК 1:
Аутофагия: механизм. Проиллюстрировано упрощенное развитие аутофагии от инициации, зародышеобразования и удлинения мембраны и созревания до окончательного слияния с лизосомой. Пути конъюгации ATG не показаны из соображений экономии места.Питательные вещества могут влиять на баланс AMPK / mTOR, чтобы модулировать аутофагию, как и клеточный стресс независимо от AMPK. Вероятно, наилучшей демонстрацией этих двух различных путей инициации в единой модели является ишемия-реперфузия, как показано. Стрелки прогрессивные, полосы — тормозящие. Маленькие треугольники, прямоугольники и овалы представляют собой груз для аутофагической деградации. Молнии и Pac-Man представляют собой лизосомальные ферменты.
РИСУНОК 1:
Аутофагия: механизм. Проиллюстрировано упрощенное развитие аутофагии от инициации, зародышеобразования и удлинения мембраны и созревания до окончательного слияния с лизосомой.Пути конъюгации ATG не показаны из соображений экономии места. Питательные вещества могут влиять на баланс AMPK / mTOR, чтобы модулировать аутофагию, как и клеточный стресс независимо от AMPK. Вероятно, наилучшей демонстрацией этих двух различных путей инициации в единой модели является ишемия-реперфузия, как показано. Стрелки прогрессивные, полосы — тормозящие. Маленькие треугольники, прямоугольники и овалы представляют собой груз для аутофагической деградации. Молнии и Pac-Man представляют собой лизосомальные ферменты.
Стрелки прогрессивные, полосы — тормозящие. Маленькие треугольники, прямоугольники и овалы представляют собой груз для аутофагической деградации. Молнии и Pac-Man представляют собой лизосомальные ферменты.
Макроаутофагия в оставшейся части этого обзора будет называться просто аутофагией. В этой форме аутофагии двойная мембранная аутофагосома сливается с лизосомным пузырьком с образованием аутолизосомы, где лизосомные протеазы могут затем получить доступ и разрушить секвестрированный груз аутофагосомы (Рисунок 1). Этот процесс является фундаментальным для избавления клетки от старых и агрегированных белков и является единственным известным механизмом удаления поврежденных органелл в нормальных условиях, а также в клетках при стрессе [15–19].Способность удалять старые и поврежденные клеточные компоненты, такие как митохондрии и эндоплазматический ретикулум (ЭР), позволяет избежать клеточного стресса, апоптоза и способствовать долголетию клеток и организма. В состояниях нормальной физиологии происходит активный аутофагический процесс, работающий во всех типах клеток почек и особенно заметный в подоцитах для защиты целостности клеток от стресса, с которым эти клетки сталкиваются на фильтрующем барьере [20].
Процесс инициации, зарождения и удлинения / созревания везикулы макроаутофагии требует нескольких различных белков AuTophaGy (ATG).Инициирование контролируется комплексом ULK1-ATG13-FIP200, для нуклеации требуется Beclin-1-Class III PI3K и связанный с ним белковый комплекс, а две системы конъюгации участвуют в удлинении, созревании или закрытии (рис. 1). Одна из этих систем конъюгации влечет за собой расщепление легкой цепи 3 (LC3) ассоциированных с микротрубочками белков до цитозольного LC3-I и дальнейшую модификацию липидирования до фосфатидилэтаноламина в мембране аутофагосомы с образованием LC3-II. Таким образом, уровни цитозольного LC3-I и связанного с аутофагосомами LC3-II являются установленными индикаторами канонической аутофагии [21].
В то время как шаперон hsc70 CMA связывает пентапептидный мотив, KFERQ, в белках, нацеленных на транслокацию в лизосому, макроаутофагия считалась неселективным процессом, захватывающим сегменты цитозоля. С тех пор были идентифицированы белки, которые избирательно усиливают аутофагосомный груз с помощью белков, нацеленных на деградацию. Лучше всего изучен линкерный белок p62 / SQSTM1 (p62). Связывающие домены на p62 включают связанный с убиквитином связывающий домен и взаимодействующую область LC3.Таким образом, p62 собирает убиквитинированные белки для деградации и связывает LC3, чтобы закрепить эти белки в формирующейся аутофагосоме. Накопление p62 в агрегаты происходит, когда аутофагия низкая или недостаточная [22]. Агрегаты p62 могут стабилизировать Nrf-2 (ядерный фактор (эритроидный 2) -подобный 2), центральный регулятор транскрипции ферментов, экспрессируемых в ответ на окислительный клеточный стресс, который может происходить при низких аутофагических состояниях [23]. Накопленный p62 также подавляет протеасомную активность [24], другое клеточное средство деградации белка.Хроническая неспособность избавиться от токсичных клеточных агрегатов еще больше увеличивает стресс и гибель клеток.
С тех пор были идентифицированы белки, которые избирательно усиливают аутофагосомный груз с помощью белков, нацеленных на деградацию. Лучше всего изучен линкерный белок p62 / SQSTM1 (p62). Связывающие домены на p62 включают связанный с убиквитином связывающий домен и взаимодействующую область LC3.Таким образом, p62 собирает убиквитинированные белки для деградации и связывает LC3, чтобы закрепить эти белки в формирующейся аутофагосоме. Накопление p62 в агрегаты происходит, когда аутофагия низкая или недостаточная [22]. Агрегаты p62 могут стабилизировать Nrf-2 (ядерный фактор (эритроидный 2) -подобный 2), центральный регулятор транскрипции ферментов, экспрессируемых в ответ на окислительный клеточный стресс, который может происходить при низких аутофагических состояниях [23]. Накопленный p62 также подавляет протеасомную активность [24], другое клеточное средство деградации белка.Хроническая неспособность избавиться от токсичных клеточных агрегатов еще больше увеличивает стресс и гибель клеток.
ПОЛОЖЕНИЕ АВТОФАГИИ
Прекрасный баланс: взаимная гармония mTOR и AMPK
Ввод питательных веществ активирует протеинкиназу млекопитающих или механистическую мишень рапамицина (mTOR). Существует два различных комплекса mTOR: Комплекс 1, где mTOR связывается с Raptor, PRAS40 и mLST8, или Комплекс 2, где он связывается с Rictor, mSIN-1, Protor-1 и mLST8.Небольшая GTPases Rag через аминокислоты или Rheb через факторы роста активирует mTORC1 и разрушает Deptor. Нижестоящие мишени индуцируют биосинтез белков и рибосом, а также рост клеток. mTORC1 подавляет аутофагию за счет фосфорилирования ULK1 и ATG13, поэтому в этом обзоре мы будем рассматривать mTORC1.
AMPK представляет собой повсеместно экспрессируемую киназу, которая играет ключевую роль в энергетическом гомеостазе. AMPK активируется в ответ на низкий расход питания или энергии, что приводит к увеличению отношения AMP (ADP) / ATP и датчика питания LKB1. Он также может активироваться адипогормонами, такими как адипонектин. Стимуляция AMPK активирует аутофагию непосредственно через фосфорилирование комплекса инициации аутофагии ULK1 (ULK1, ATG13, FIP200) в другом сайте, чем mTORC1, а также подавляет активность mTORC1 посредством фосфорилирования TSC2, что поддерживает Rheb в неактивном состоянии. Ингибирование mTORC1 также способствует аутофагии. Таким образом, AMPK подавляет процессы расходования энергии, такие как синтез белка, а также обеспечивает защиту митохондрий и биогенез [25], экспрессию клеточных белков антиоксидантной защиты [26], подавление белков внеклеточного матрикса [27] и индукцию eNOS [ 28], но подавление iNOS [29].Для большинства клеток существует баланс между mTOR, который вызывает анаболические процессы использования энергии, и AMPK / аутофагией, которые способствуют катаболическим процессам сохранения энергии.
Он также может активироваться адипогормонами, такими как адипонектин. Стимуляция AMPK активирует аутофагию непосредственно через фосфорилирование комплекса инициации аутофагии ULK1 (ULK1, ATG13, FIP200) в другом сайте, чем mTORC1, а также подавляет активность mTORC1 посредством фосфорилирования TSC2, что поддерживает Rheb в неактивном состоянии. Ингибирование mTORC1 также способствует аутофагии. Таким образом, AMPK подавляет процессы расходования энергии, такие как синтез белка, а также обеспечивает защиту митохондрий и биогенез [25], экспрессию клеточных белков антиоксидантной защиты [26], подавление белков внеклеточного матрикса [27] и индукцию eNOS [ 28], но подавление iNOS [29].Для большинства клеток существует баланс между mTOR, который вызывает анаболические процессы использования энергии, и AMPK / аутофагией, которые способствуют катаболическим процессам сохранения энергии.
В периоды, когда пищи много, активность AMPK снижается, позволяя последующему mTORC1 увеличиваться. Клетки и организмы будут синтезировать новые белки и жирные кислоты и увеличивать метаболическую активность. Когда потребление пищи ограничено, активация AMPK действует, чтобы сохранить расход энергии, использовать внутриклеточные источники энергии и расщеплять старые и поврежденные белки и органеллы с помощью аутофагии, чтобы генерировать строительные блоки для будущей индукции mTOR анаболических процессов [30].Биоритм этих противоположных биологических циклов необходим для долголетия. При ишемии-реперфузии (IR) в сердце [31] и почках (неопубликованные данные) индукция AMPK происходит во время ишемической фазы дефицита питательных веществ и снижается при реперфузии, где индуцируется mTOR. Пожалуйста, обратитесь к Рисунку 1 для схематического представления взаимодействия между AMPK и mTOR в индукции аутофагии. Sirt1 играет роль в активации аутофагии путем деацетилирования аутофагических факторов, включая ATG5, ATG7 и LC3 [32].Активация Sirt1 увеличением соотношения NAD +: NADH в ответ на упражнения или голодание может быть отнесена к AMPK.
Существует неразрывная взаимосвязь между анаболическим, mTOR, и катаболическим, аутофагическим путями. Индукция AMPK / аутофагии приводит к катаболической деградации клеточных компонентов с образованием аминокислот. Затем аминокислоты составляют часть цикла, активирующего анаболический путь mTORC1 через Rag (рис. 2). Чтобы приспособиться к секреторному фенотипу в стареющих клетках, эта обратная связь может быть нарушена посредством чего оба пути активируются посредством реорганизации эндомембранной системы, называемой компартментом пространственного связывания TOR-аутофагии (TASCC) [33].В TASCC зрелые аутофагосомы сливаются с лизосомами, связанными с mTOR. Аминокислоты, генерируемые аутофагией, используются для активации mTOR и рекрутирования его на лизосомальную мембрану и, таким образом, способствуют синтезу секретируемых белков, характерных для стареющих клеток. В этом процессе mTOR остается пространственно отличным от ULK1 и образования ранних аутофагосом, позволяя прогрессировать как аутофагии, так и синтезу белка. TASCC наблюдалась в подоцитах и может способствовать синтезу белка в этих клетках, которые поддерживают высокую конститутивную аутофагию.Линкерный белок p62 является еще одним связующим звеном катаболического / анаболического цикла. Он также связывает Raptor и необходим для стимуляции mTORC1 в ответ на аминокислоты [34]. Таким образом, функция p62 служит не только как селективный линкер груза для аутофагии, где он разрушается под действием аутофагии, но также контролирует аутофагию, стимулируя активность mTORC1 в присутствии аминокислот, подавляя тем самым его собственную деградацию. Индукция mTORC1 необходима для биогенеза лизосом [35] и, таким образом, поддерживает аутофагию.Дефицит p62 увеличивает активность адипогенного регулятора PPARγ, а также придает характеристики метаболического синдрома непереносимости глюкозы, инсулинорезистентности, воспаления, снижения расхода энергии и ожирения у зрелых мышей [36].
РИСУНОК 2:
Аутофагия: регулирование. Показана линейная диаграмма со стрелками в виде прогрессивных путей и блокирующими полосами. Синие линии в первую очередь катаболические, а черные — анаболические. Чрезмерная активность mTORC1 может привести к увеличению белков, росту, адипогенезу и липогенезу.В сочетании с ингибированием аутофагии mTORC1 для контроля качества и удаления дисфункциональных белков и органелл прогрессирование может привести к клеточному стрессу. Эта диаграмма также иллюстрирует систему сдержек и противовесов между катаболическим и анаболическим путями, результат, если один путь, mTORC1 в соответствии с диабетом, искажает этот баланс, и зависимость одного пути от другого (т. Е. Аминокислот, генерируемых аутофагией для активации mTORC1, и поколение лизосом mTORC1 поддерживает аутофагию).
Показана линейная диаграмма со стрелками в виде прогрессивных путей и блокирующими полосами. Синие линии в первую очередь катаболические, а черные — анаболические. Чрезмерная активность mTORC1 может привести к увеличению белков, росту, адипогенезу и липогенезу.В сочетании с ингибированием аутофагии mTORC1 для контроля качества и удаления дисфункциональных белков и органелл прогрессирование может привести к клеточному стрессу. Эта диаграмма также иллюстрирует систему сдержек и противовесов между катаболическим и анаболическим путями, результат, если один путь, mTORC1 в соответствии с диабетом, искажает этот баланс, и зависимость одного пути от другого (т. Е. Аминокислот, генерируемых аутофагией для активации mTORC1, и поколение лизосом mTORC1 поддерживает аутофагию).
РИСУНОК 2:
Аутофагия: регулирование.Показана линейная диаграмма со стрелками в виде прогрессивных путей и блокирующими полосами. Синие линии в первую очередь катаболические, а черные — анаболические. Чрезмерная активность mTORC1 может привести к увеличению белков, росту, адипогенезу и липогенезу. В сочетании с ингибированием аутофагии mTORC1 для контроля качества и удаления дисфункциональных белков и органелл прогрессирование может привести к клеточному стрессу. Эта диаграмма также иллюстрирует систему сдержек и противовесов между катаболическим и анаболическим путями, результат, если один путь, mTORC1 согласно диабету, искажает этот баланс, и зависимость одного пути от другого (т.е. аминокислоты, образующиеся в результате аутофагии для активации mTORC1, и генерация лизосом mTORC1, поддерживающая аутофагию).
Правильный баланс этих путей обеспечивает баланс питания для правильного гомеостаза клеток и долголетия. Текущие исследования показывают, что для снижения стресса и сохранения здоровья клеток требуется баланс между регуляцией этих путей. Этот цикл аутофагической деградации, образования аминокислот, индукции mTORC1 и лизосомной регенерации устанавливает маятниковую взаимосвязь между анаболическим и катаболическим циклами и их зависимость друг от друга. Это хроническое ингибирование или активация катаболической, аутофагической или анаболической, mTORC1, сторон цикла отрицательно влияет на весь цикл (рис. 2). Это можно оценить по нарушению этого биоритма при заболевании или по использованию фармакологических или генетических средств для нарушения этого баланса и имитации заболевания, как будет описано ниже. Для дальнейшей поддержки этого биоритма белок аутофагии ATG14 регулируется циркадным ритмом [37]. Следовательно, для восстановления баланса фармацевтические препараты следует вводить с учетом этого цикла.
Это хроническое ингибирование или активация катаболической, аутофагической или анаболической, mTORC1, сторон цикла отрицательно влияет на весь цикл (рис. 2). Это можно оценить по нарушению этого биоритма при заболевании или по использованию фармакологических или генетических средств для нарушения этого баланса и имитации заболевания, как будет описано ниже. Для дальнейшей поддержки этого биоритма белок аутофагии ATG14 регулируется циркадным ритмом [37]. Следовательно, для восстановления баланса фармацевтические препараты следует вводить с учетом этого цикла.
Митофагия в ответ на клеточный стресс
Существуют разные формы аутофагии в ответ на разные сигнальные пути, и мы только сейчас начинаем понимать их различия и последствия [38]. Как показано на Рисунке 1, хотя ишемия снижает количество питательных веществ и активирует AMPK, реперфузия увеличивает клеточный стресс, который вызывает аутофагию независимо от статуса питательных веществ. Клеточный стресс может повредить митохондрии и ER, что приведет к образованию ROS, а также окисленных и неправильно свернутых белков.Аутофагический ответ может избирательно нацеливаться, приобретать и разрушать эти органеллы и затронутые белки. Особый интерес представляют механизмы, участвующие в избирательной митохондриальной макроаутофагии или митофагии.
Митохондрии широко считаются клеточными энергетическими установками с аэробной выработкой АТФ, с другими функциями, включая передачу сигналов кальция и апоптоз. Потребность в клетках и физиологические условия играют роль в количестве и размере этих органелл. Например, для выполнения транспортных функций проксимального канальца почек, требующих высокой энергии, требуется большое количество митохондрий.Избирательная деградация происходит за счет аутофагии в сочетании с событиями деления / слияния. В болезненных состояниях этот механизм может стать дополнительным налогом, чтобы избежать гибели клеток. Поврежденные или состаренные митохондрии демонстрируют пониженный потенциал митохондриальной мембраны, который активирует предполагаемую киназу 1 (PINK1), индуцированную PTEN, и E3-убиквитинлигазу, Parkin. PINK1 конститутивно продуцируется и связывает митохондриальную мембрану, но имеет очень короткий период полураспада. Повреждение митохондрий стабилизирует белки PINK1, которые накапливаются на внешней митохондриальной мембране и привлекают паркин.Комплекс PINK1 / Parkin выполняет двойную функцию убиквитинирования как митофузинов для предотвращения слияния митохондрий, так и потенциал-зависимого анионного канала 1. Эти полиубиквитинированные митохондрии связывают линкерный белок p62, а затем LC3 на развивающейся аутофагосоме, что приводит к секвестрации и удалению митохондрий. с помощью аутофагии. Текущий обзор митофагии см. В [39].
Поврежденные или состаренные митохондрии демонстрируют пониженный потенциал митохондриальной мембраны, который активирует предполагаемую киназу 1 (PINK1), индуцированную PTEN, и E3-убиквитинлигазу, Parkin. PINK1 конститутивно продуцируется и связывает митохондриальную мембрану, но имеет очень короткий период полураспада. Повреждение митохондрий стабилизирует белки PINK1, которые накапливаются на внешней митохондриальной мембране и привлекают паркин.Комплекс PINK1 / Parkin выполняет двойную функцию убиквитинирования как митофузинов для предотвращения слияния митохондрий, так и потенциал-зависимого анионного канала 1. Эти полиубиквитинированные митохондрии связывают линкерный белок p62, а затем LC3 на развивающейся аутофагосоме, что приводит к секвестрации и удалению митохондрий. с помощью аутофагии. Текущий обзор митофагии см. В [39].
Запрограммированный процесс удаления митохондрий во время развития, как в красных кровяных тельцах, демонстрирует участие только Bh4-белков Nix (также известного как Bnip3L) и Bnip3.Эти митохондриально-связывающие белки способны связываться с аутофагосомой LC3 напрямую, не требуя линкерного мостика убиквитин-p62. Ранее было показано, что этот механизм реагирует на стрессовую стимуляцию и вызывает апоптоз или некроз. Сейчас считается, что этот механизм используется для поддержания исходного митохондриального гомеостаза посредством аутофагии, но может регулировать митохондриальную аутофагию, апоптоз или некроз в зависимости от величины и продолжительности клеточного стресса [40]. Гипоксия также может индуцировать Bnip3, который может вытеснять Beclin1 из Bcl-2 и способствовать аутофагии [41].
ER unfolded protein response (UPR) — это путь клеточной реакции на стресс, задействованный для восстановления гомеостаза ER после стресса или старения, нарушающих функцию ER. ER UPR использует протеасомные и аутофагические механизмы, чтобы избавиться от неправильно свернутых белков и поврежденных ER. ER стресс клеток проксимальных канальцев почек увеличивает реакцию аутофагии [42]. Оба пути UPR PERK [43] и IRE1 [44] индуцируют аутофагию. Подобно митофагии, аутофагия ER или ретикулофагия, по-видимому, является механизмом защиты клеток.АФК могут быть общим фактором индукции клеточного стресса механизмов аутофагического ответа [45].
Оба пути UPR PERK [43] и IRE1 [44] индуцируют аутофагию. Подобно митофагии, аутофагия ER или ретикулофагия, по-видимому, является механизмом защиты клеток.АФК могут быть общим фактором индукции клеточного стресса механизмов аутофагического ответа [45].
Липофагия
Аутофагия не только служит механизмом гомеостаза и контроля качества клеточного стресса, но также генерирует энергию для клетки в то время, когда количество питательных веществ мало. Энергия, получаемая из аминокислот, относительно неэффективна, но недавно было показано, что аутофагия способствует более энергоэффективному катаболизму внутриклеточных липидных капель (LD) [46] или липофагии.Почему существуют две системы, способные мобилизовать LD, аутофагию и цитозольные липазы, не известно и требует дальнейшего изучения.
Голодание увеличивает базальную липофагию для выработки энергии, в то время как липидный вызов также увеличивает базальную липофагию и приводит к избирательной секвестрации LD. Однако хроническая липидная нагрузка приводит к увеличению LD в клетке [46]. Такое расширение области LD, наблюдаемое при 4-месячной диете с высоким содержанием жиров (HFD), связано с прогрессирующей неспособностью аутофагосомы сливаться с лизосомой (Рисунок 3) [47].По-видимому, виновником является более высокий состав холестерина в аутофагосоме, а истощение холестерина в клетках фибробластов увеличивает аутофагию [48]. Таким образом, изменение липидного состава мембран, по-видимому, является причиной неспособности слияния аутофагосомы и лизосомы, что, следовательно, превращает увеличение аутофагии в ответ на HFD на подавление аутофагии в долгосрочной перспективе [47].
РИСУНОК 3:
Липофагия. При избыточной липидной нагрузке или при недостатке питательных веществ аутофагия используется для мобилизации LD.В первом случае липофагия предназначена для поддержания способности / гомеостаза LD, а во втором — для получения энергии. Хроническое введение липидов может в конечном итоге ингибировать слияние аутофагосом и лизосом, вызывая накопление везикул аутофагосом.
Хроническое введение липидов может в конечном итоге ингибировать слияние аутофагосом и лизосом, вызывая накопление везикул аутофагосом.
РИСУНОК 3:
Липофагия. При избыточной липидной нагрузке или при недостатке питательных веществ аутофагия используется для мобилизации LD. В первом случае липофагия предназначена для поддержания способности / гомеостаза LD, а во втором — для получения энергии. Хроническое введение липидов может в конечном итоге ингибировать слияние аутофагосом и лизосом, вызывая накопление везикул аутофагосом.
Аутофагия, влияющая на HFD и ожирение: ссылка на инсулинорезистентность
Исследования, изучающие влияние HFD на аутофагию и влияние аутофагии на орган, проводились в основном в гипоталамусе, печени, сердце, β-клетках поджелудочной железы и адипоцитах. Ожирение и HFD отрицательно влияют на баланс и реакцию аутофагии и связанных с ней путей.
Жировые клетки участвуют в мобилизации LD не только для снабжения энергией в периоды недостаточного снабжения питательными веществами, но также для предотвращения увеличения запасов от нарушения целостности клеток в ответ на большую липидную нагрузку.В соответствии с этим объяснением, HFD или ожирение приводит к увеличению аутофагии в адипоцитах, и в исследовании пациентов с ожирением аутофагия была особенно повышена в жировой ткани сальника [49]. В адипоцитах пациентов с ожирением и диабетом 2 типа (T2D) наблюдается снижение mTORC1 с одновременным увеличением аутофагии, нарушением митохондрий, стрессом ER и цитозольной LD [50]. Адипонектин — это адипокин, который уменьшается при ожирении. Было показано, что он имеет отрицательную корреляцию с альбуминурией у пациентов с ожирением, а мыши с дефицитом адипонектина демонстрируют повышенную альбуминурию и слитные отростки стопы подоцитов [10].Повышенный уровень аутофагии в жировой ткани в ответ на вызванный ожирением ER стресс увеличивает деградацию и, таким образом, снижает уровни адипонектина [51], а также рецепторов инсулина [52]. Таким образом, предполагалось, что повышенная аутофагическая деградация играет роль в резистентности к инсулину. Исследования мышей с нокаутом гена аутофагии ATG7 подтвердили эту концепцию. У этих мышей с нарушенной аутофагией были обнаружены меньшие адипоциты с мультиокулярными LD, увеличенное количество митохондрий и, как следствие, скорость β-окисления.То есть эти адипоциты напоминали коричневую жировую ткань [53]. Кроме того, мыши были более стройными, более энергичными и устойчивыми к индуцированному HFD ожирению и инсулинорезистентности [53, 54]. У пациентов с ожирением T2D неспособность инсулина активировать mTORC1 в адипоцитах способствует сверхактивной аутофагии [50].
Таким образом, предполагалось, что повышенная аутофагическая деградация играет роль в резистентности к инсулину. Исследования мышей с нокаутом гена аутофагии ATG7 подтвердили эту концепцию. У этих мышей с нарушенной аутофагией были обнаружены меньшие адипоциты с мультиокулярными LD, увеличенное количество митохондрий и, как следствие, скорость β-окисления.То есть эти адипоциты напоминали коричневую жировую ткань [53]. Кроме того, мыши были более стройными, более энергичными и устойчивыми к индуцированному HFD ожирению и инсулинорезистентности [53, 54]. У пациентов с ожирением T2D неспособность инсулина активировать mTORC1 в адипоцитах способствует сверхактивной аутофагии [50].
В отличие от адипоцитов, печень демонстрирует снижение аутофагии и увеличение ER стресса у мышей с ожирением (ob / ob) [55]. Снижение уровня ATG7 для подавления аутофагии даже у худых мышей достаточно, чтобы вызвать стресс ER и инсулинорезистентность.Кроме того, восстановление аутофагии посредством аденовирусной экспрессии ATG7 у мышей ob / ob увеличивало чувствительность к инсулину и снижало стресс ER у этих животных по сравнению с мышами ob / ob с контрольным вектором. Хроническое кормление HFD нарушает способность аутофагосом сливаться с лизосомами в печени [47]. Актуальность аутофагии при ожирении была далее продемонстрирована на β-клетках поджелудочной железы, где избирательное подавление аутофагии путем скрещивания мышей с дефицитом ATG7, специфичных к β-клеткам, с мышами ob / ob приводило к усилению стресса ER, гибели β-клеток и тяжелому диабету [56]. .Β-клетки поджелудочной железы также обнаруживают накопление аутофагосом и снижение экспрессии генов лизосом в ответ на HFD или диабет, что указывает на низкий аутофагический поток [57]. HFD в гипоталамусе также приводит к дефекту аутофагии, который коррелирует с усилением воспаления, стрессом ER и ожирением [58]. HFD-индукция аутофагии в жировой ткани вредна, но, по-видимому, не распространяется на эти другие органы, где аутофагия оказалась полезной. Однако прогрессирование дефектной аутофагии в этих органах может привести к осложнениям, нивелирующим защитные свойства.
Однако прогрессирование дефектной аутофагии в этих органах может привести к осложнениям, нивелирующим защитные свойства.
АУТОФАГИЯ ПРИ ХБН, СВЯЗАННЫХ С ОЖИРЕНИЕМ
Несмотря на то, что с HFD и аутофагией в почках было проведено мало работы, были проведены обширные исследования по моделям почечного диабета и текущие обзоры на тему аутофагии почек и диабета, а также других заболеваний почек [59–61].
Старение может способствовать прогрессированию многих заболеваний, включая ХБП. Как активность AMPK, так и аутофагия снижаются с возрастом, тогда как старение приводит к увеличению холестерина, свободных жирных кислот, ROS и в ответ на повышенный клеточный стресс переход к фенотипу старения.У стареющих мышей со специфическим для подоцитов дефицитом ATG5 наблюдается повышенный стресс ER, накопление окисленных и убиквитинированных белков и органелл, протеинурия и повреждение клубочков [20]. Подоциты играют ключевую роль в поддержании фильтрационного барьера, который, по-видимому, является уязвимым компонентом почек, поскольку повреждение подоцитов может привести к протеинурии и гломерулосклерозу, основным признакам диабетической нефропатии. Еще больше усложняет проблему то, что эти клетки являются постмитотическими с ограниченной пролиферативной способностью и должны управлять клеточными стрессами в сильно изменчивой среде.По этим причинам в этих клетках может потребоваться высокая конститутивная аутофагическая активность для подавления клеточного стресса и поддержания целостности подоцитов.
Этот высокий уровень аутофагии, низкий статус mTORC1, типичный для подоцитов, трансформируется в низкий уровень аутофагии, высокий статус mTORC1 при многих болезненных состояниях, включая диабетическую нефропатию. Это может привести к накоплению поврежденных и убиквитинированных белков и органелл, которые увеличивают клеточный стресс и гипертрофию подоцитов, повреждение и потерю подоцитов и, в конечном итоге, гломерулярное заболевание с прогрессирующей протеинурией и гломерулосклерозом [62]. Чтобы еще больше проиллюстрировать этот баланс, Inoki et al . [63] гиперактивированный селективный по подоцитам mTORC1 у мышей и наблюдались повреждения подоцитов и гломерулярные поражения, сходные с диабетическим фенотипом, включая протеинурию и мезангиальное расширение. Эти данные в значительной степени согласуются с выводами Hartleben et al . [20] стареющих мышей с дефицитом подоцитов ATG5, упомянутым выше, за исключением гиперактивных подоцитов мышей mTORC1, также демонстрирующих избыточный рост подоцитов. Напротив, ингибирование mTORC1 рапамицином в значительной степени подавляет эти осложнения и, следовательно, диабетический фенотип [64, 65].Частично это может быть связано с реактивацией гомеостатической аутофагии. При назначении рапамицина пациентам действительно возникали проблемы, в первую очередь увеличение протеинурии, связанное с подоцитами [66]. Считалось, что при длительном воздействии рапамицин может влиять на mTORC2 и актиновые компоненты цитоскелета, необходимые для структуры щелевой диафрагмы подоцитов, и действительно, щелочная диафрагма и связанные с ней белки подавляются рапамицином [67]. Интересно, что делеция Raptor (специфичного для mTORC1) сама по себе также приводит к повреждению клубочков [68], что указывает на более сложную парадигму.Недавно было показано, что ингибирование mTOR в подоцитах подавляет образование новых лизосом, что приводит к накоплению аутофагосом [69]. Это еще раз иллюстрирует взаимозависимость катаболических путей аутофагии с анаболическими путями mTOR (Рисунок 2).
Чтобы еще больше проиллюстрировать этот баланс, Inoki et al . [63] гиперактивированный селективный по подоцитам mTORC1 у мышей и наблюдались повреждения подоцитов и гломерулярные поражения, сходные с диабетическим фенотипом, включая протеинурию и мезангиальное расширение. Эти данные в значительной степени согласуются с выводами Hartleben et al . [20] стареющих мышей с дефицитом подоцитов ATG5, упомянутым выше, за исключением гиперактивных подоцитов мышей mTORC1, также демонстрирующих избыточный рост подоцитов. Напротив, ингибирование mTORC1 рапамицином в значительной степени подавляет эти осложнения и, следовательно, диабетический фенотип [64, 65].Частично это может быть связано с реактивацией гомеостатической аутофагии. При назначении рапамицина пациентам действительно возникали проблемы, в первую очередь увеличение протеинурии, связанное с подоцитами [66]. Считалось, что при длительном воздействии рапамицин может влиять на mTORC2 и актиновые компоненты цитоскелета, необходимые для структуры щелевой диафрагмы подоцитов, и действительно, щелочная диафрагма и связанные с ней белки подавляются рапамицином [67]. Интересно, что делеция Raptor (специфичного для mTORC1) сама по себе также приводит к повреждению клубочков [68], что указывает на более сложную парадигму.Недавно было показано, что ингибирование mTOR в подоцитах подавляет образование новых лизосом, что приводит к накоплению аутофагосом [69]. Это еще раз иллюстрирует взаимозависимость катаболических путей аутофагии с анаболическими путями mTOR (Рисунок 2).
РЕЗЮМЕ
Аутофагия тщательно сбалансирована с циклами анаболического питания и необходима для клеточного гомеостаза и продолжительности оргазма. Из-за естественного циркадного ритма этого уравновешивающего действия селективные агонисты или антагонисты также следует вводить соответствующим образом.Аутофагия также может реагировать на клеточные и экологические стрессоры в краткосрочной перспективе, но в долгосрочной перспективе постоянная потребность в таких селективных и защитных действиях может нарушить нормальные гомеостатические процессы расщепления старых и поврежденных белков и органелл для детоксикации клетки и образования новые сотовые строительные блоки. HFD и ожирение отрицательно влияют на баланс и реакцию этих путей. То, что начинается как защитный механизм разрушения LD в ответ на липидную нагрузку, у животных, находящихся на хронической липофагии HFD, становится вредным для адипоцитов.Аналогичная проблема может возникнуть при диабете, когда подоциты переходят от высокой аутофагии, низкого статуса mTOR к низкой аутофагии, высокому статусу mTOR. Прямое ингибирование mTOR с помощью фармакологических или генетических средств может оказаться полезным на животных моделях, но из-за взаимозависимости этих путей такое вмешательство может в конечном итоге негативно повлиять на аутофагию и увеличить клеточный стресс в долгосрочной перспективе. Пациенты также могут быть более чувствительны к этим терапевтическим стратегиям, если у них уже есть проблемы со здоровьем.В целом понимание взаимосвязи этих путей проливает свет на благотворное влияние диеты и физических упражнений на пациентов с ожирением и диабетом.
Другие формы аутофагии, включая CMA [13], пероксисомную селективную аутофагию, пексофагию [70], аутофагию ER, ретикулофагию, а также несколько других белков и путей, таких как Sirt1 [71], могут влиять на катаболический анаболический баланс, но не обсуждались из-за ограничений по длине. Прошу прощения у коллег, чьи работы не цитировались из-за тех же ограничений.
ЗАЯВЛЕНИЕ О КОНФЛИКТЕ ИНТЕРЕСОВ
Не заявлено
БЛАГОДАРНОСТЬ
Наши проекты поддерживаются грантами от VA MERIT Award (KS) и NIDDK (награды U01 DK060995, DP3 DK094352-01 и DK083142) в KS, Национальные институты здравоохранения Гранты DK56248, DK28602, UAB-UCSD Центр острых заболеваний О’Брайена Грант на исследование травм почек P30DK079337 (JS) и Департамент по делам ветеранов, VA Merit Awards 5101 и 0011, Управление здравоохранения ветеранов, Управление исследований и разработок.
ССЫЛКИ
1« и др.
Распространенность избыточной массы тела и ожирения в США, 1999–2004 гг.
,
JAMA
,
2006
, vol.
295
(стр.
1549
—
1555
) 2« и др.
Ожирение и риск хронической почечной недостаточности
,
J Am Soc Nephrol
,
2006
, vol.
17
(стр.
1695
—
1702
) 3« и др.
Факторы риска терминальной стадии почечной недостаточности: 25-летнее наблюдение
,
Arch Intern Med
,
2009
, vol.
169
(стр.
342
—
350
) 4« и др.
Эпидемиология ожирения
,
Гастроэнтерология
,
2007
, т.
132
(стр.
2087
—
2102
) 5« и др.
Связь между ожирением и заболеванием почек: систематический обзор и метаанализ
,
Kidney Int
,
2008
, vol.
73
(стр.
19
—
33
) 6« и др.
Профилактика заболеваний почек, связанных с ожирением: возрастные эффекты ограничения диетического питания
,
Kidney Int
,
2002
, vol.
62
(стр.
208
—
219
) 7,,, et al.
Функциональные и структурные изменения почек на ранних стадиях ожирения
,
J Am Soc Nephrol
,
2001
, vol.
12
(стр.
1211
—
1217
) 8« и др.
Похудание и протеинурия: систематический обзор клинических испытаний и сравнительных когорт
,
Nephrol Dial Transplant
,
2010
, vol.
25
(стр.
1173
—
1183
) 9.
Ожирение и хроническая болезнь почек
,
Нефрология
,
2011
, т.
31
(стр.
397
—
403
) 10,,, et al.
Адипонектин регулирует альбуминурию и функцию подоцитов у мышей
,
J Clin Invest
,
2008
, vol.
118
(стр.
1645
—
1656
) 11,.
Опосредованная шапероном аутофагия в контроле качества белка
,
Curr Opin Cell Biol
,
2011
, vol.
23
(стр.
184
—
189
) 12.
Рост почки при катаболической болезни: то, что она не разрушает, делает ее сильнее
,
J Ren Nutr
,
2007
, vol.
17
(стр.
167
—
172
) 13« и др.
Подавление опосредованной шапероном аутофагии в коре почек при остром сахарном диабете
,
Kidney Int
,
2004
, vol.
65
(стр.
2135
—
2144
) 14,,.
Микроаутофагия: малоизвестное самопоедание
,
Cell Mol Life Sci
,
2012
, vol.
69
(стр.
1125
—
1136
) 15,.
Recycle or die: роль аутофагии в кардиопротекции
,
J Mol Cell Cardiol
,
2008
, vol.
44
(стр.
654
—
661
) 16,.
Аутофагия: основные принципы и отношение к болезни
,
Annu Rev Pathol
,
2008
, vol.
3
(стр.
427
—
455
) 17,.
Аутофагия при старении, болезни и смерти: истинная личность самозванца клеточной смерти
,
Cell Death Differ
,
2009
, vol.
16
(стр.
1
—
2
) 18,,, et al.
Аутофагия борется с болезнями посредством клеточного самопереваривания
,
Nature
,
2008
, vol.
451
(стр.
1069
—
1075
) 19,.
Аутофагия: защитный механизм против повреждения почек, вызванного нефротоксикантами
,
Kidney Int
,
2009
, vol.
75
(стр.
118
—
119
) 20,,, et al.
Аутофагия влияет на восприимчивость к заболеваниям клубочков и поддерживает гомеостаз подоцитов у стареющих мышей
,
J Clin Invest
,
2010
, vol.
120
(стр.
1084
—
1096
) 21« и др.
LC3, гомолог дрожжевого Apg8p у млекопитающих, локализуется в мембранах аутофагосом после обработки
,
EMBO J
,
2000
, vol.
19
(стр.
5720
—
5728
) 22« и др.
Гомеостатические уровни p62 контролируют образование цитоплазматических телец включения у мышей с дефицитом аутофагии
,
Cell
,
2007
, vol.
131
(стр.
1149
—
1163
) 23« и др.
Селективный субстрат аутофагии p62 активирует стресс-чувствительный фактор транскрипции Nrf2 посредством инактивации Keap1
,
Nat Cell Biol
,
2010
, vol.
12
(стр.
213
—
223
) 24« и др.
Ингибирование аутофагии нарушает деградацию субстратов убиквитин-протеасомного пути
,
Mol Cell
,
2009
, vol.
33
(стр.
517
—
527
) 25« и др.
Повышающая регуляция митохондриального разобщающего протеина-2 с помощью AMP-активированной протеинкиназы в эндотелиальных клетках снижает окислительный стресс при диабете
,
Диабет
,
2008
, vol.
57
(стр.
3222
—
3230
) 26,.
AMPKalpha1 регулирует антиоксидантный статус эндотелиальных клеток сосудов
,
Biochem J
,
2009
, vol.
421
(стр.
163
—
169
) 27,,, et al.
AMP-активированная протеинкиназа ингибирует Smad3-зависимую транскрипцию и трансдифференцировку миофибробластов, индуцированную трансформирующим фактором роста
,
J Biol Chem
,
2008
, vol.
283
(стр.
10461
—
10469
) 28« и др.
Прямая активация АМФ-активированной протеинкиназы стимулирует синтез оксида азота в эндотелиальных клетках аорты человека
,
J Biol Chem
,
2003
, vol.
278
(стр.
31629
—
31639
) 29,,.
Ресвератрол Ингибирование индуцибельной синтазы оксида азота в скелетных мышцах включает AMPK, но не SIRT1
,
Am J Physiol Endocrinol Metab
,
2011
, vol.
301
(стр.
E922
—
E930
) 30.
Гены аутофагии и старение
,
Cell Death Differ
,
2009
, vol.
16
(стр.
94
—
102
) 31« и др.
Отличительные роли аутофагии в сердце во время ишемии и реперфузии: роль AMP-активированной протеинкиназы и Beclin 1 в опосредовании аутофагии
,
Circ Res
,
2007
, vol.
100
(стр.
914
—
922
) 32« и др.
Роль НАД-зависимой деацетилазы Sirt1 в регуляции аутофагии
,
Proc Natl Acad Sci USA
,
2008
, vol.
105
(стр.
3374
—
3379
) 33« и др.
Пространственная связь mTOR и аутофагии увеличивает секреторные фенотипы
,
Science
,
2011
, vol.
332
(стр.
966
—
970
) 34« и др.
p62 является ключевым регулятором восприятия питательных веществ в пути mTORC1
,
Mol Cell
,
2011
, vol.
44
(стр.
134
—
146
) 35,,, et al.
Прекращение аутофагии и реформация лизосом, регулируемая mTOR
,
Nature
,
2010
, vol.
465
(стр.
942
—
946
) 36« и др.
Ожирение в зрелом возрасте и инсулинорезистентность у мышей с дефицитом сигнального адаптера p62
,
Cell Metab
,
2006
, vol.
3
(стр.
211
—
222
) 37,,, et al.
Ген 14, связанный с аутофагией (Atg14), регулируется факторами транскрипции Forkhead Box O и циркадными ритмами и играет важную роль в аутофагии печени и метаболизме липидов
,
J Biol Chem
,
2012
, vol.
287
(стр.
39107
—
39114
) 38,.
Аутофагия: адаптируемый модификатор онкогенеза
,
Curr Opin Genet Dev
,
2010
, vol.
20
(стр.
57
—
64
) 39,.
Механизмы митофагии
,
Nat Rev Mol Cell Biol
,
2011
, vol.
12
(стр.
9
—
14
) 40.
Bnip3 как двойной регулятор митохондриального обмена и гибели клеток в миокарде
,
Pediatr Cardiol
,
2011
, vol.
32
(стр.
267
—
274
) 41« и др.
Аутофагия, индуцированная гипоксией, опосредуется индуцируемым гипоксией фактором индукции BNIP3 и BNIP3L через их домены Bh4
,
Mol Cell Biol
,
2009
, vol.
29
(стр.
2570
—
2581
) 42« и др.
Стресс эндоплазматического ретикулума вызывает аутофагию в клетках проксимальных канальцев почек
,
Трансплантат Nephrol Dial
,
2009
, vol.
24
(стр.
2665
—
2672
) 43« и др.
ER стресс (фосфорилирование PERK / eIF2alpha) опосредует индуцированное полиглутамином превращение LC3, важный этап для формирования аутофагии
,
Cell Death Differ
,
2007
, vol.
14
(стр.
230
—
239
) 44« и др.
Аутофагия активируется для выживания клеток после стресса эндоплазматического ретикулума
,
Mol Cell Biol
,
2006
, vol.
26
(стр.
9220
—
9231
) 45,.
Регуляция аутофагии с помощью АФК: физиология и патология
,
Trends Biochem Sci
,
2011
, vol.
36
(стр.
30
—
38
) 46« и др.
Аутофагия регулирует липидный обмен
,
Nature
,
2009
, vol.
458
(стр.
1131
—
1135
) 47,,.
Измененное содержание липидов подавляет аутофагическое слияние пузырьков
,
FASEB J
,
2010
, vol.
24
(стр.
3052
—
3065
) 48« и др.
Истощение холестерина вызывает аутофагию
,
Biochem Biophys Res Commun
,
2006
, vol.
351
(стр.
246
—
252
) 49« и др.
Измененная аутофагия в жировой ткани человека при ожирении
,
J Clin Endocrinol Metab
,
2011
, vol.
96
(стр.
E268
—
E277
) 50« и др.
Ослабление передачи сигналов mTOR и усиление аутофагии в адипоцитах пациентов с ожирением и диабетом 2 типа
,
Mol Med
,
2010
, vol.
16
(стр.
235
—
246
) 51,.
Аутофагия: роль в вызванном ожирением стрессе ER и подавлении адипонектина в адипоцитах
,
Аутофагия
,
2010
, vol.
6
(стр.
1196
—
1197
) 52« и др.
Опосредованное аутофагией подавление рецепторов инсулина способствует индуцированной стрессом эндоплазматической сети инсулинорезистентности
,
Mol Pharmacol
,
2009
, vol.
76
(стр.
596
—
603
) 53« и др.
Жировоспецифическая делеция гена 7, связанного с аутофагией (atg7), у мышей выявляет роль в адипогенезе
,
Proc Natl Acad Sci USA
,
2009
, vol.
106
(стр.
19860
—
19865
) 54,,.
Аутофагия и адипогенез: влияние на ожирение и диабет II типа
,
Аутофагия
,
2010
, т.
6
(стр.
179
—
181
) 55,,, et al.
Нарушение аутофагии печени при ожирении способствует стрессу ER и вызывает инсулинорезистентность
,
Cell Metab
,
2010
, vol.
11
(стр.
467
—
478
) 56« и др.
Дефицит аутофагии в бета-клетках приводит к нарушению реакции развернутого белка и прогрессированию от ожирения к диабету у мышей
,
Diabetologia
,
2012
, vol.
55
(стр.
392
—
403
) 57« и др.
Жирные кислоты подавляют аутофагический оборот в бета-клетках
,
J Biol Chem
,
2011
, vol.
286
(стр.
42534
—
42544
) 58,.
Дефектная гипоталамическая аутофагия направляет центральный патогенез ожирения через путь IkappaB киназы бета (IKKbeta) / NF-kappaB
,
J Biol Chem
,
2011
, vol.
286
(стр.
32324
—
32332
) 59« и др.
Возрастающая роль аутофагии в функции почек, заболеваниях и старении
,
Аутофагия
,
2012
, т.
8
(стр.
1009
—
1031
) 60« и др.
Аутофагия: новая терапевтическая мишень при заболеваниях почек
,
Clin Exp Nephrol
,
2012
, vol.
16
(стр.
827
—
832
) 61« и др.
Аутофагия как терапевтическая мишень при диабетической нефропатии
,
Exp Diabetes Res
,
2012
, vol.
2012
стр.
628978
62,.
Влияние аутофагии на старение и болезни клубочков
,
Cell Tissue Res
,
2011
, vol.
343
(стр.
467
—
473
) 63« и др. Активация
mTORC1 в подоцитах является критическим этапом в развитии диабетической нефропатии у мышей
,
J Clin Invest
,
2011
, vol.
121
(стр.
2181
—
2196
) 64« и др.
Путь mTOR высокоактивирован при диабетической нефропатии, и рапамицин обладает сильным терапевтическим потенциалом
,
Biochem Biophys Res Commun
,
2009
, vol.
384
(стр.
471
—
475
) 65,,, et al.
Рапамицин предотвращает ранние стадии развития диабетической нефропатии у крыс
,
Am J Nephrol
,
2007
, vol.
27
(стр.
495
—
502
) 66« и др.
Рапамицин оказывает двойное противоположное действие на протеинурические экспериментальные нефропатии: связано ли это с повреждением подоцитов?
,
Циферблат нефрола
,
2009
, т.
24
(стр.
3632
—
3640
) 67« и др.
Сиролимус и протеинурия у пациентов с трансплантатом почки: доказательства дозозависимого эффекта на белки, связанные с щелевой диафрагмой
,
Трансплантация
,
2011
, vol.
91
(стр.
997
—
1004
) 68« и др.
Роль mTOR в функции подоцитов и диабетической нефропатии у людей и мышей
,
J Clin Invest
,
2011
, vol.
121
(стр.
2197
—
2209
) 69« и др.
Ингибирование MTOR нарушает аутофагический поток в подоцитах
,
J Am Soc Nephrol
,
2012
, vol.
23
(стр.
412
—
420
) 70« и др.
Пексофагия: избирательная деградация пероксисом
,
Int J Cell Biol
,
2012
, vol.
2012
стр.
512721
71« и др.
Сиртуины и почечные заболевания: связь со старением и диабетической нефропатией
,
Clin Sci (Lond)
,
2013
, vol.
124
(стр.
153
—
164
)
© Автор, 2013. Опубликовано Oxford University Press от имени ERA-EDTA. Все права защищены.
Нарушение метаболизма и фиброз жировой ткани: роль коллагена VI
Жировая ткань является ключевым регулятором системного энергетического гомеостаза. Физиологическое состояние жировой ткани определяется клеточно-автономными процессами внутри адипоцита. В дополнение к этому сам адипоцит подвергается серьезным модификациям со стороны других типов клеток, которые проникают в жировую ткань, таких как макрофаги и сосудистые клетки; кроме того, на адипоциты могут заметно влиять некоторые гормоны и цитокины, которые циркулируют в системной системе.
Хотя все эти клеточные взаимодействия были предметом обширных исследований в многочисленных лабораториях, внеклеточный матрикс жировой ткани на сегодняшний день получил ограниченное внимание, несмотря на данные, свидетельствующие о том, что это функционально значимая составляющая физиологии жировой ткани.
В настоящее время неизвестно, какие последствия метаболический стресс оказывает на внеклеточный матрикс и наоборот. Другими словами, как влияет нарушение регуляции внеклеточных компонентов жировой ткани на системное метаболическое состояние? Здесь мы подходим к этому вопросу с двух разных точек зрения.Сначала мы оценили общий уровень компонентов внеклеточного матрикса в различных метаболических условиях и установили, что внеклеточные компоненты глобально активируются во время метаболически сложных условий. Затем мы выбрали конкретного члена семейства коллагена, коллаген VI (демонстрирующий преобладающую экспрессию в жировой ткани), и использовали генетическую модель разрушения коллагена VI для исследования эффектов разрушения внеклеточного матрикса жировой ткани. Примечательно, что наши исследования продемонстрировали, что такое ослабление внеклеточного матрикса жировой ткани приводит к значительному улучшению метаболического фенотипа в контексте как диеты с высоким содержанием жиров, так и проблемы с мутацией ob / ob .
Наши наблюдения подчеркивают, что внеклеточный матрикс жировой ткани является важным и новым местом модуляции системного метаболизма. При ожирении жировая ткань имеет признаки, сходные с признаками других фиброзных тканей, таких как печень; это предполагает, что определенные компоненты этой обычно довольно жесткой среды внеклеточного матрикса могут служить возможными мишенями для фармакологического вмешательства с целью лечения метаболических нарушений.
МАТЕРИАЛЫ И МЕТОДЫ
Животные.Все протоколы экспериментов на животных были одобрены Институтом исследований на животных Медицинского колледжа Альберта Эйнштейна или Комитетом по уходу и использованию животных при Юго-Западном медицинском центре Техасского университета в Далласе. Мышей с нулевым уровнем коллагена VI получали, как описано ранее Bonaldo с коллегами (4). Мыши с нулевым уровнем коллагена VI ob / ob ( col6KOob / ob ) были получены путем скрещивания гетерозиготных мышей с нулевым уровнем коллагена VI (фон FVB) с гетерозиготными мышами с дефицитом лептина ( ob / + ) (фон C57).Мышей содержали группами от двух до пяти в клетках с верхними фильтрами. Колония содержалась в свободном от патогенов учреждении, аккредитованном AALAC, в Медицинском колледже Альберта Эйнштейна в условиях контролируемой среды (от 22 до 25 ° C; от 40 до 50% влажности). Как указано, мышей поддерживали на 12-часовом цикле света и темноты с неограниченным доступом к воде и стандартной пищевой диете (№ 5058; Lab-Diet) или диете с высоким содержанием жиров (№ D12492; Research Diets Inc.). Эксперименты с мышами с нулевым уровнем коллагена VI проводились на чистом фоне FVB, а с мышами col6KOob / ob — на смешанном фоне C57 / BL6 и FVB.Все эксперименты контролировались однопометниками. Большинство экспериментов с мышами col6KOob / ob также независимо воспроизводились на чистом фоне FVB.
Люди. Это исследование было одобрено институциональным наблюдательным советом Юго-Западного медицинского центра Юта (Даллас, Техас). Письменное информированное согласие было получено от каждого участника. Участников набирали с помощью публичных рекламных объявлений (листовок), размещенных в колледжах, церквях, храмах и продуктовых магазинах Южной Азии в метро Даллас-Форт-Уэрт.Рост и вес измеряли стандартными методами. Подготовленный технический персонал провел опросник по общему состоянию здоровья. Диабет был исключен путем измерения уровней глюкозы и глюкозы в плазме натощак во время стандартного перорального теста на толерантность к глюкозе. Биопсию жировой ткани получали после голодания в течение ночи с использованием иглы для биопсии Temno II 14 калибра 9 см (Allegiance) для взятия пробы из подкожных областей живота и ягодичных (периферических) областей. После обработки кожи бетадином на брюшной стенке был сделан небольшой разрез кожи с помощью скальпеля №11.Это облегчило ведение биопсийной иглы в жировое подкожное пространство. Жир собирали с брюшной стенки в правом нижнем квадранте на 2 см выше и медиальнее переднего бугорка подвздошной кости и из ягодичной области.
Пероральный тест толерантности к глюкозе и тест толерантности к инсулину. Для перорального теста толерантности к глюкозе мышей не кормили за 2 часа до введения глюкозы (2,5 г / кг массы тела [BW]) через желудочный зонд. Хвостовую венозную кровь собирали и анализировали на глюкозу и инсулин.Глюкозу измеряли с помощью анализа оксидаза-пероксидаза (Sigma-Aldrich), а инсулин измеряли с помощью набора для иммуноферментного анализа инсулина (Millipore, Billerica, MA). В ходе исследования мышам было отказано в доступе к пище. Для теста на толерантность к инсулину мышей не кормили за 2 часа до введения 1 ед. Человеческого инсулина (Novo Nordisk) / кг массы тела путем внутрибрюшинной инъекции. Хвостовую венозную кровь собирали и анализировали на глюкозу.
Клиренс триглицеридов. Мышей не кормили в течение 2 часов и вводили 15 мкл оливкового масла на грамм веса тела через желудочный зонд.Приблизительно 20 мкл крови собирали в каждый момент времени и анализировали на триглицериды (набор триглицеридов Infinity; Thermo Electron Corp.) и свободные жирные кислоты (FFA) (NEFA C; Wako). В ходе исследования мышам было отказано в доступе к пище.
Заражение LPS. Самцам мышей в возрасте 10 недель внутрибрюшинно вводили липополисахарид (LPS; Sigma-Aldrich) в дозе 0,1 мг / кг массы тела. Что касается временного графика провокации LPS, кровь собирали через 0, 3, 6 и 24 часа и анализировали на интерлейкин-6 (IL-6) и моноцитарный хемоаттрактантный белок 1 (MCP-1; R&D Systems), как указано.Для исследования специфического воспаления жировой ткани в ответ на ЛПС мышей умерщвляли через 90 минут после инъекции ЛПС, а ткани немедленно замораживали в жидком азоте.
желудочный зонд для агониста PPARγ. Агонист 2- (2- [4-фенокси-2-пропилфенокси] этил) индол-5-уксусная кислота (COOH) был любезным подарком от Merck Research Laboratories. (Рэуэй, Нью-Джерси). COOH вводили самцам мышей FVB в возрасте 10 недель через желудочный зонд ежедневно в 12 часов дня в течение 14 дней в дозе 10 мг / кг массы тела.Мышей умерщвляли через 6 часов после последнего желудочного зондирования, и ткани немедленно замораживали в жидком азоте.
Передача сигналов инсулина in vivo. Мышей не кормили в течение 2 часов и внутрибрюшинно вводили человеческий инсулин в дозе 1 Ед / кг МТ. Мышей умерщвляли через 0, 5 или 10 мин после инъекции, и ткани немедленно замораживали в жидком азоте. Ткани гомогенизировали в буфере для анализа радиоиммунопреципитации, дополненном полным коктейлем ингибиторов протеазы и коктейлем ингибиторов фосфатазы (Roche).Лизат белка непосредственно анализировали вестерн-блоттингом с антифосфо-AKT (Cell Signaling Technology Inc.) и анти-Akt-1 (Santa Cruz Biotechnology Inc.). Вестерн-блоттинг выполняли с использованием системы инфракрасной визуализации Odyssey (LI-COR Biotechnology).
Иммуноблот-анализ. Жировые ткани гомогенизировали в буфере TNET без Triton X-100 (150 мМ NaCl, 5 мМ EDTA, 50 мМ Tris-HCl, pH 7,5) с добавлением полного коктейля ингибиторов протеазы и коктейля ингибиторов фосфатазы (Roche).После этого следовало центрифугирование на низкой скорости (3000 × г, при 4 ° C), чтобы удалить жировую лепешку с верхней части пробирки. Тритон Х-100 добавляли к гомогенату до конечной концентрации 1%, и экстракт очищали при 20000 × г в течение 15 мин при 4 ° C и смешивали с 2 × буфером для образцов Лэммли. Пробы белков разделяли на 4–12% бис-трис-гелях с последующим переносом на нитроцеллюлозу ВА83. Как указано, блоты зондировали различными антителами. Киназа, регулируемая фосфо-внеклеточным сигналом (фосфо-ERK), фосфо-JNK, фосфо-p38, ERK и антитела JNK были получены от Santa Cruz Biotechnology; антитело р38 было получено от Biosource; фосфо-SMAD2, фосфо-SMAD3 и общие антитела SMAD3 были получены от Cell Signaling; общее антитело SMAD2 было получено от Invitrogen.Связанные антитела выявляли с помощью конъюгированных с IRDye800 антикроличьих антител или конъюгированных с IRDye700 вторичных антител против мышиных (Rockland). Мембраны сканировали с помощью системы инфракрасной визуализации LI-COR Odyssey, и интенсивность полос количественно определяли с помощью программного обеспечения Odyssey v1.2 (LI-COR Biotechnology).
Профилирование экспрессии генов. Животных умерщвляли удушением CO 2 через 6 часов после последней дозы (для лечения лекарственными средствами), собирали белую жировую ткань эпидидимии и быстро замораживали в жидком азоте.Полную РНК выделяли после гомогенизации замороженных тканей в реагенте TRIzol (Invitrogen) с помощью PT10 / 35 Polytron (Kinematica AG) и обрабатывали с использованием наборов RNeasy (Qiagen) в соответствии с инструкциями производителей. Концентрации РНК оценивали по оптической плотности при 260 нм. Обработку микрочипов выполняли, как описано ранее (24). Вкратце, меченую кРНК гибридизовали в течение 48 ч на 60-мерных двухцветных микроматрицах Agilent с пятнами. Индивидуальные образцы для обработки штамма или соединения (включая индивидуальный контроль или образцы для обработки носителем) гибридизовали с контрольным пулом или пулом для обработки носителем.Изменения и значения P были определены путем усреднения повторов (от двух до трех повторов на штамм или обработку) и с использованием программного обеспечения Rosetta Resolver v6.0 (Rosetta Inpharmatics LLC, дочерняя компания Merck & Co., Inc., Сиэтл , Вашингтон). Значения изменений представляют разницу в регулировании образцов, обработанных штаммом или соединением, по сравнению с контрольным образцом или образцом, обработанным носителем, а положительное значение означает усиление регуляции после обработки соединением или наоборот.
Количественный анализ ПЦР в реальном времени.Мышей умерщвляли, ткани немедленно собирали и замораживали в жидком азоте. РНК экстрагировали из ткани с помощью реагента TRIzol (Invitrogen) с последующим выделением РНК с помощью набора для ткани Qiagen RNeasy. Общую РНК (1 мкг) подвергали обратной транскрипции с помощью обратной транскриптазы SuperScript III и олиго (dT) 20 (Invitrogen). Количественная ПЦР в реальном времени (qRT-PCR) была проведена с использованием мастер-микса Sybr Green I. Ее проводили в Roche Lightcycler 480. Праймеры были сконструированы таким образом, чтобы охватить интрон, чтобы предотвратить амплификацию любой загрязняющей геномной ДНК, если она присутствует.Все ПЦР были нормализованы до 18S рРНК, если не указано иное, и относительные уровни экспрессии были определены методом ΔΔ C t с эффективностью всех праймеров, равной ~ 2 (35), что позволило нам провести сравнение относительной численности разные коллагены. Используемые наборы праймеров перечислены в таблице S2 дополнительного материала.
Иммуногистохимия и процедуры окрашивания. Свежую жировую ткань фиксировали в 10% забуференном фосфатом формалине в течение ночи.Срезы парафинового воска размером 5 мкм обрабатывали для иммуноокрашивания. Для иммуногистохимического окрашивания срезы обрабатывали буфером для инактивации пероксидазы в течение 10 минут при комнатной температуре для подавления эндогенной пероксидазной активности (Dako) и инкубировали с первичными антителами в течение 24 часов при 4 ° C. После отмывки срезы инкубировали с биотинилированными вторичными антителами в течение 1 ч при комнатной температуре и проявляли реакцию с помощью реагента ABC (Vector Laboratories). Все слайды контрастировали гематоксилином (Sigma-Aldrich).Окрашивание F4 / 80 проводили с помощью моноклонального антитела крысы (Invitrogen) и биотинилированного вторичного антитела кролика против крысы (Vector Laboratories). Дополнительные методы окрашивания включали подкожные срезы кожи, окрашенные трихромом Массона и гематоксилином и эозином (H&E). Окрашивание концевой дезоксинуклеотидилтрансферазы dUTP-биотином по нику-концам (TUNEL) проводили в соответствии с протоколом производителя (Chemicon). Все изображения были получены с помощью охлаждаемой камеры Censys с заряженной связью (Photometrics, Tucson, AZ) на аксиофото Zeiss (Zeiss, Йена, Германия).
Количественное определение размера адипоцитов. Срезы придатковой и брыжеечной жировой ткани 10-недельных мышей окрашивали H&E. Программное обеспечение Image J использовалось для измерения площади адипоцитов, которая представлена как средняя площадь адипоцитов (в мкм 2 ) или, альтернативно, как процент адипоцитов в каждой 100-мкм площади 2 . Размер адипоцитов измеряли у четырех мышей / генотип (> 500 клеток / генотип).
Иммунофлуоресцентное окрашивание инсулина / глюкагона и количественное определение размера островков.Ткань поджелудочной железы фиксировали фиксатором Буэна (насыщенная пикриновая кислота-формальдегид-ледяная уксусная кислота в соотношении 15: 5: 1) в течение 5 часов. Парафиновые срезы (5 мкм) инкубировали с контрольным иммуноглобулином G осла (1: 500; Jackson Immunoresearch Laboratories Inc.) в течение 1 ч для блокирования неспецифического связывания. Затем срезы инкубировали с антителами морской свинки к инсулину мыши (1: 500; любезный подарок от Регины Кулиават, Медицинский колледж Альберта Эйнштейна, Бронкс, штат Нью-Йорк) и кроличьими антителами против глюкагона человека (1: 500; Invitrogen) в течение ночи при 4 ° С.После трехкратной промывки 1 × фосфатно-солевым буфером срезы инкубировали с конъюгированными с флуоресцеином изотиоцианатом антителами осла против морских свинок и конъюгированными с Texas Red антителами осла против кроликов (1: 250; Jackson Immunoresearch Laboratories Inc.) в течение 1 часа. при комнатной температуре. Окрашенные H и E срезы всей поджелудочной железы вырезали так, чтобы была видна вся поверхность ткани, и использовали для количественного определения размера островков. Островки визуализировались при 10-кратном увеличении; площадь островка измерялась с помощью изображения J и выражалась как относительная площадь островка / общая площадь сечения поджелудочной железы, как описано в ссылке 47.Срезы анализировали с помощью микроскопа Olympus IX81.
TEM и SEM. Для просвечивающей электронной микроскопии (TEM) свежую жировую ткань разрезали на 1 мкм 2 кусочков и фиксировали в течение ночи в 2% параформальдегиде, 2,5% глутаральдегиде в 0,1 М буфере какодилата натрия. Их постфиксировали 1% тетроксидом осмия, а затем 1% уранилацетатом, дегидратировали с помощью градиентной серии этанола и заключали в смолу LX112 (LADD Research Industries, Burlington, VT). Ультратонкие срезы (80 нм) вырезали на приборе Reichert Ultracut UCT, окрашивали уранилацетатом, а затем цитратом свинца и просматривали на просвечивающем электронном микроскопе JEOL 1200EX при 80 кВ.Для сканирующей электронной микроскопии (SEM) свежую жировую ткань разрезали на небольшие блоки и анализировали, как описано в ссылке 7. Вкратце, ткань фиксировали в течение ночи фиксатором Карновского 1: 1 с последующим применением метода OTOTO. Образцы обезвоживали в этаноле с постепенным изменением концентрации и пропитывали гексаметилдисилазаном. Образцы закрепляли на алюминиевых штырях и исследовали в сканирующем электронном микроскопе JEOL JSM6400 (Пибоди, Массачусетс) с использованием ускоряющего напряжения 10 кВ. Ложная окраска была добавлена к изображениям с помощью Adobe Photoshop.
Содержание коллагена. Содержание коллагена в тканях определяли путем окрашивания фиксированных формалином парафиновых срезов эпидидимальной жировой ткани в пикросириус красный (Sigma-Aldrich). Измерение гидроксипролина проводилось с использованием модифицированного протокола, описанного в другом месте (55). Вкратце, 100 мг замороженного жира нагревали в 6 н. HCl при 110 ° C в течение ночи в запаянных пробирках. Затем образцы нагревали при 110 ° C в течение 48 ч до высыхания. Каждый образец инкубировали с хлорамином-Т (Sigma-Aldrich) при комнатной температуре точно в течение 20 минут, а затем с p -диметиламинобензальдегид (Fisher Scientific) при 60 ° C в течение 15 минут.Поглощение считывали при 540 нм, а концентрацию определяли по стандартной кривой, построенной для цис -4-гидрокси-1-пролина (Sigma-Aldrich).
Измерения триглицеридов печени. Триглицериды экстрагировали из замороженных тканей печени (200 мг), как описано Carr et al. (6а). Триглицериды измеряли колориметрическим анализом с использованием триглицеридов Infinity (Thermo Scientific).
Состав тела и измерения косвенной калориметрии Состав тела измеряли с помощью магнитно-резонансной томографии (МРТ) с использованием аппарата Echo MRI (Echo Medical Systems, Хьюстон, Техас).Для непрямых калориметрических измерений животные были индивидуально помещены в метаболические камеры, поддерживаемые при температуре от 20 до 22 ° C в течение 12 часов / 12 часов цикла свет-темнота с включением света в 07:00. Метаболические измерения (потребление кислорода, коэффициент респираторного обмена и двигательная активность). активности) были получены непрерывно с использованием системы непрямой калориметрии с открытым контуром CLAMS (Columbus Instruments, Columbus, OH). Мышам давали стандартный рацион, упомянутый выше, и воду из-под крана ad libitum. Представленные результаты содержат данные, собранные за период 8 дней после 3 дней адаптации к метаболическим клеткам.Для измерения потребления пищи мышей содержали отдельно и взвешивали пищу каждый день в полдень в течение 7 дней. Потребление пищи выражается в граммах съеденной пищи на грамм веса тела.
Статистический анализ. Результаты представлены как средние значения ± стандартная ошибка среднего. Статистический анализ проводили с помощью теста Стьюдента t или с помощью двухфакторного дисперсионного анализа (ANOVA) и последующего теста Тьюки с использованием GraphPad Prism 5 (Сан-Диего, Калифорния). Значимость была принята при значении P <0.05.
РЕЗУЛЬТАТЫ
Коллагены сильно активируются в жировой ткани во время метаболических проблем. Чтобы оценить уровень внеклеточных компонентов при различных метаболических условиях, мы выполнили крупномасштабное профилирование экспрессии генов на жировой ткани нескольких моделей мышей, у которых метаболические состояния варьируются в широком диапазоне. Сравнение микроматрицы проводили на эпидидимальных жировых подушечках у мышей db / db и мышей дикого типа (WT). Параллельно мы сравнивали мышей WT, получавших антидиабетический агонист PPARγ (COOH), с мышами, получавшими носитель.Мы также распространили анализ на трансгенных мышей, которые постоянно сверхэкспрессируют адипонектин из адипоцитов (adipoTG), на мышиной модели, которая, как мы недавно продемонстрировали, имеет увеличенную жировую массу с метаболически здоровым фенотипом, имитирующим хроническое воздействие агонистов PPARγ (15, 28). Результаты микроматрицы показали, что подавляющее большинство коллагенов активируется в жировой ткани у мышей db / db (рис. 1A). Напротив, мыши, получавшие COOH, или мыши с хронической сверхэкспрессией адипонектина демонстрируют подавление коллагенов, которые активируются в состоянии db / db , что позволяет предположить, что большинство компонентов внеклеточного матрикса являются либо прямыми, либо косвенными мишенями для PPARγ.Поскольку все обычные коллагены (за исключением коллагена IX) ведут себя на удивление однородно и активируются при диабетическом состоянии, мы полагаем, что термин «фиброз жировой ткани» точно описывает повышенное присутствие компонентов внеклеточного матрикса во время состояний ожирения и воспаления. Этот повышенный фиброз жировой ткани может привести к повышенной жесткости жировой ткани, потенциально ограничивая дальнейшее разрастание отдельных жировых клеток. Чтобы лучше понять относительное содержание различных коллагенов, мы выполнили qRT-PCR с обычными коллагенами от I до VI (рис.1B) в нормальной жировой подушке дикого типа. Коллагены I, IV и VI обильно экспрессировались в зрелых адипоцитах, что согласуется с отчетами из других лабораторий (39), в то время как коллагены II и III имели значительно более низкие уровни экспрессии в адипоцитах. Однако коллаген VI был наиболее преобладающим коллагеном во всех исследованных жировых депо.
РИС. 1. Уровни экспрессии коллагена VI
коррелируют с различными состояниями метаболизма. (A) Профили экспрессии генов коллагенов, экспрессируемых в основном в жировой ткани, на моделях мышей с метаболической нагрузкой выполняли с помощью анализа микроматриц.Все анализы микрочипов выполнялись с использованием эпидидимального жира. Результаты выражены как изменение экспрессии гена в дб / дб по сравнению с мышами WT (верхний график), изменение у мышей WT, получавших агонист PPARγ (30 мг / кг COOH в течение 7 дней), по сравнению с мышами WT, получавшими носитель (средний график) и изменение у мышей со сверхэкспрессией адипонектина по сравнению с мышами WT (нижний график). Меченую кРНК из каждого индивидуального экспериментального штамма и обработки ( n ≥ трех мышей) конкурентно гибридизовали против пула кРНК из соответствующей контрольной группы ( n ≥ трех мышей) на микрочипах Agilent.Положительное изменение свидетельствует об усилении регуляции в экспериментальном штамме или группе лечения, и наоборот. (B) Коллаген VI — это коллаген, который преимущественно экспрессируется в жире. Основные экспрессируемые в жировой ткани коллагены (коллаген I, II, III, IV, V и VI) измеряли в эпидидимальных, брыжеечных, паховых и околопочечных жировых подушках с помощью qRT-RCR у 8-недельных мышей WT ( n = 2). Уровни экспрессии нормализовали до 18S рРНК. Показан типичный пример. (C) Экспрессия коллагена VI увеличивается в жировой ткани.Уровни коллагена VI альфа 3 измеряли с помощью qRT-PCR в основных жировых подушках и других тканях 8-недельных мышей WT ( n = 2). Уровни экспрессии нормализовали до 18S рРНК. Показан типичный пример. (D) Уровни экспрессии коллагена VI альфа 3 положительно коррелируют с метаболически затрудненными состояниями. Уровни экспрессии определяли с использованием микрочипов Agilent, как описано для панели A. Результаты показывают изменение уровней экспрессии коллагена VI альфа 3 у мышей ob / ob и db / db по сравнению с мышами WT и изменение у мышей WT, получавших 10 или 30 мг / кг (mpk) агониста PPARγ по сравнению с обработкой носителем (верхняя панель) ( n ≥ трех мышей / группа, за исключением обработки агонистом PPARγ 10 мг / кг, которая включала двух мышей).Экспрессия коллагена VI была снижена в жировой ткани обработкой PPARγ (средняя и нижняя панели). Мышам дикого типа (возраст 10 недель) вводили 30 мг / кг COOH или носитель в течение 14 дней через желудочный зонд. Уровни мРНК коллагена VI альфа 1, альфа 2 и альфа 3 измеряли в придатном (средняя панель) и брыжеечном (нижняя панель) жирах с помощью qRT-PCR и нормализовали до 18S рРНК (среднее значение ± стандартная ошибка; n = четыре мыши. /группа). (E) Экспрессия коллагена VI повышена у лиц с метаболическим ожирением.Уровни коллагена VI альфа 3 были измерены в подкожной брюшной и ягодичной (периферической) жировой ткани у азиатских индейцев (18 мужчин и 4 женщины), группы с метаболическим ожирением, и сравнивались с контрольными европейскими популяциями того же возраста (27 ± 4 и 28 ± 7). лет соответственно) и индексов массы тела (24 ± 4 и 25 ± 6 кг / м 2 соответственно). Уровни экспрессии измеряли с помощью qRT-PCR и нормализовали до 18S рРНК (средние значения ± стандартные ошибки; n = от 15 до 19 кавказцев, n = от 24 до 30 азиатов).*, P <0,05; **, P <0,01 по критерию Стьюдента t .
Экспрессия коллагена VI увеличивается в жировой ткани. В соответствии с нашими первоначальными наблюдениями, мы идентифицировали коллаген VI альфа 3 ( col6a3 ) как главный секреторный белок адипоцитов (48) и впоследствии продемонстрировали, что отсутствие жировой ткани — производный коллаген значительно снижает рост опухоли в молочной железе (26). Коллаген VI отличается от других членов семейства коллагенов тем, что он существенно обогащен жировой тканью (рис.1С). Результаты этих экспериментов подтвердили обогащение коллагена VI в жировой ткани, демонстрируя высокие уровни экспрессии в эпидидимальных, брыжеечных, подкожных и околопочечных жировых подушках по сравнению с уровнями, обнаруженными в других тканях. Следовательно, коллаген VI может играть важную роль в физиологии адипоцитов; следовательно, коллаген VI был выбран для дальнейшего углубленного анализа.
Профили экспрессии гена эпидидимального жира у мышей ob / ob и db / db продемонстрировали значительно повышенные уровни col6a3 во время состояний метаболического стресса с активацией 1.3-кратное и 1,4-кратное соответственно. Напротив, обработка мышей дикого типа агонистом PPARγ значительно снижала уровни col6a3 в 1,6–1,9 раза (фиг. 1D, верхняя панель также см. Таблицу S1 в дополнительном материале). Исследование нескольких отдельных жировых подушечек с помощью qRT-PCR показало, что 14-дневная обработка агонистом PPARγ снижает уровни всех трех альфа-цепей коллагена VI в 1,4-1,5 раза в придатковидном жире (рис. 1D, средняя панель) и В 1,6–2,1 раза в мезентериальном жире (рис. 1D, нижняя панель).
Чтобы установить, ограничен ли этот феномен повышения уровня коллагена VI во время периодов метаболической нагрузки грызунами или он также актуален в контексте заболеваний человека, мы исследовали уровни col6a3 в популяции азиатских индейцев и сравнил их с подобранной контрольной группой кавказцев. Несмотря на схожие индексы массы тела, азиатское население Индии имеет более высокий уровень восприимчивости к инсулинорезистентности, чем европейское население (10).Частично это происходит из-за дисфункциональной жировой ткани, которая имеет тенденцию к большему воспалению, даже после поправки на подкожную жировую ткань между азиатскими индейцами и европейцами (9). Таким образом, это предоставило уникальную возможность отделить ожирение от метаболической дисфункции. В соответствии с наблюдениями на доклинических моделях с метаболической нагрузкой, экспрессия col6a3 была значительно увеличена как в брюшной, так и в ягодичной подкожной жировой клетчатке в этой азиатской индийской когорте (рис.1E). Это говорит о том, что повышенная регуляция коллагена VI также является признаком дисрегуляции жировой ткани человека.
ob / ob мыши, лишенные коллагена VI, имеют меньшую массу тела в раннем возрасте. Чтобы выяснить, коррелирует ли повышенная жесткость внеклеточного матрикса только с метаболически затрудненным состоянием, или это движущая сила, оказывающая прямое функциональное влияние на жировые отложения ткани, мы исследовали метаболические последствия дестабилизации внеклеточного матрикса на генетической модели мыши.Коллаген VI представляет собой гетеротример, собранный из трех альфа-цепей: альфа 1, альфа 2 и альфа 3. Как описано ранее (4), нарушение коллагена VI альфа 1 приводит к общему функциональному нулевому фенотипу для коллагена VI, поскольку в отсутствие цепи альфа 1, цепи альфа 2 и 3 не могут эффективно секретироваться. Это приводит к отсутствию отложения коллагена VI во внеклеточном матриксе. Мы подвергли мышей с нулевым уровнем коллагена VI ( col6KO ) сильной метаболической нагрузке путем скрещивания этих нулевых мышей с фоном ob / ob .Анализ увеличения веса в зависимости от возраста показал, что мыши col6KOob / ob имели меньшую массу тела в раннем возрасте и оставались легче примерно до 11-недельного возраста. После этого момента, вес тела мышей col6KOob / ob догнал их однопометников ob / ob , и их вес больше не отличался статистически (рис. 2A). Анализ состава тела с помощью эхо-МРТ показал, что различия в весе, наблюдаемые в более молодом возрасте, в основном связаны с различиями в массе жира, тогда как у более старых животных состав тела сопоставим с мышами ob / ob (рис.2Б).
РИС. 2.
У мышей col6KOob / ob уменьшилась масса тела из-за уменьшения жировой массы в молодом возрасте. (A) Вес тела контролировали в течение 10 недель (среднее значение ± стандартная ошибка; n = четыре мыши / группа). (B) Состав тела 8-недельных и 12-недельных мышей определяли с помощью магнитно-резонансной томографии с использованием ECHO MRI (среднее значение ± стандартная ошибка; n = четыре мыши / группа). *, P <0,05. (A) Данные были проанализированы с помощью двухфакторного дисперсионного анализа; (B) данные были проанализированы с помощью теста Стьюдента t .
Недостаток коллагена VI улучшает метаболический фенотип у мышей ob / ob . Несколько метаболических параметров были измерены у мышей col6KOob / ob . Уровни глюкозы натощак у 10-недельного ребенка col6KOob / ob нормализовались (рис. 3А). При проведении перорального теста на толерантность к глюкозе мыши col6KOob / ob показали резко улучшенный клиренс глюкозы в течение всего периода заражения. Сопоставимые метаболические улучшения были также обнаружены у 12-недельных мышей, когда различия в ожирении исчезли (рис.3Б). Аналогичные тенденции наблюдались, если мыши col6KO без мутации ob / ob были подвергнуты диете с высоким содержанием жиров. Недостаток коллагена VI приводит к снижению прибавки в весе (рис. 3C) из-за снижения накопления жира (репрезентативная картина подкожного и внутрикожного жировых слоев показана на рис. 3D) с улучшением клиренса глюкозы во время пероральной толерантности к глюкозе. тест (данные не показаны). Поскольку фенотип, полученный в результате воздействия диеты с высоким содержанием жиров, и фенотип мышей col6KO , скрещенных с фоном ob / ob , были почти идентичными, мы выполнили оставшиеся метаболические характеристики с использованием мышей, несущих мутацию ob / ob .
РИС. 3.
Отсутствие коллагена VI нормализует метаболический профиль у мышей ob / ob и мышей, получавших провокационную пищу с высоким содержанием жиров. (A и B) Уровень глюкозы натощак (слева) и уровни циркулирующей глюкозы во время перорального теста на толерантность к глюкозе (справа) у 10-недельного (A) и 12-недельного (B) ребенка col6KOob / ob и ob / ob мышей. (C) Вес тела контролировали у однопометников с нулевым уровнем коллагена VI и дикого типа. 12-недельная диета с высоким содержанием жиров была начата на 9-й неделе. На панелях от А до С данные представлены в виде средних значений ± стандартные ошибки ( n = четыре мыши / группа).(D) Окрашивание трихромом по Массону подкожных срезов кожи мышей с нулевым уровнем коллагена VI и мышей WT после 12-недельного провокационного испытания на диете с высоким содержанием жиров (HFD). (E) Циркулирующие триглицериды (Trig) (левая панель) и FFA (правая панель) были измерены во время липидной провокации у 10-недельных мышей col6KOob / ob (среднее значение ± стандартная ошибка; n = четыре мыши / группа) . (F) Уровни триглицеридов после еды и натощак у 10-недельных мышей col6KOob / ob . (G) Содержание триглицеридов в печени измеряли экстракцией хлороформ-метанол и колориметрическим анализом (как описано в разделе «Материалы и методы») (средние значения ± стандартные ошибки; n = четыре мыши / группа).(H) H & E-окрашенные срезы поджелудочной железы от 10-недельных мышей-самцов показали снижение гиперплазии островков у мышей col6KOob / ob по сравнению с ob / ob однопометников (верхняя панель). Срезы полной поверхности поджелудочной железы использовались для количественного определения площади островков. Площадь островков определяли при 10-кратном увеличении и выражали как площадь островков (мкм 2 ) / площадь всего сечения поджелудочной железы (мкм 2 ) (средние значения ± стандартные ошибки; n = пять мышей / группа). Иммунофлуоресцентное окрашивание срезов поджелудочной железы на инсулин (зеленый) и глюкагон (красный) показало улучшение распределения α- и β-клеток у мышей col6KOob / ob (нижняя панель).(I) Просвечивающая электронная микроскопия эпидидимальной жировой ткани от 16-недельных мышей col6KOob / ob и ob / ob . Стрелка указывает на типичных инвагинированных кавеол, а острие стрелки указывает на невагинированные кавеолы. Двуглавая стрелка указывает интерстициальное пространство между двумя адипоцитами. (J) Передача сигналов инсулина у 10-недельных мышей col6KOob / ob и их однопометников ob / ob , которым вводили 1 ЕД инсулина / кг массы тела и у которых собирали эпидидимальную жировую ткань через 0, 5 и 10 мин. постинъекция.Состояние фосфорилирования AKT определяли вестерн-блоттингом непосредственно со специфическими антителами к P-AKT и общим уровням AKT. Интенсивность полос количественно оценивали с помощью системы визуализации LI-COR (LI-COR Biosciences) (средние значения ± стандартные ошибки; n = три мыши / группа). *, P <0,05; **, P <0,01 (по критерию Стьюдента t или с помощью двустороннего дисперсионного анализа).
У мышей col6KOob / ob наблюдается улучшение клиренса липидов. Дополнительные улучшения у мышей col6KOob / ob наблюдались во время перорального введения триглицеридов.В отличие от пониженной скорости клиренса липидов, которая обычно наблюдается у мышей ob / ob , мыши col6KOob / ob имели повышенную скорость удаления триглицеридов (рис. 3E, слева). Однако не было значительного изменения сывороточных FFA во время этой липидной провокации (фиг. 3E, справа). Уровни триглицеридов натощак и после еды оставались неизменными у мышей col6KOob / ob (фиг. 3F). Мыши col6KOob / ob также показали значительное снижение степени накопления липидов в печени.Мыши ob / ob накапливали характерно высокие уровни триглицеридов печени (рис. 3G), тогда как уровни липидов печени у мышей col6KOob / ob были значительно улучшены. Эти результаты предполагают, что в отсутствие коллагена VI у мышей не только улучшается углеводный обмен, но также наблюдается значительное улучшение липидного обмена.
Недостаток коллагена VI улучшает гиперплазию поджелудочной железы, типичную для мышей ob / ob .Принимая во внимание многочисленные улучшения углеводного и липидного метаболизма в отсутствие коллагена VI, возникает вопрос, могут ли эти улучшения проявиться в дальнейшем на уровне поджелудочной железы. Мы гистологически исследовали островки поджелудочной железы, уделяя особое внимание β-клеткам. Значительное улучшение морфологии островков поджелудочной железы действительно может наблюдаться у мышей col6KOob / ob . Окрашивание H&E поджелудочной железы выявило ожидаемую гиперплазию β-клеток, характерную для островков ob / ob по сравнению с островками дикого типа, что вызвано высокой степенью инсулинорезистентности (рис.3H, верхняя панель). Недостаток коллагена VI привел к уменьшению площади островков. Это также стало очевидным после более количественной оценки. Иммунофлуоресцентный анализ островков был выполнен для дальнейшего подчеркивания улучшений в общей архитектуре островков путем окрашивания на α (красные) и β (зеленые) клетки, чтобы выделить количество и распределение этих двух типов клеток в островках (рис. 3H, нижняя панель). Островки от мышей col6KOob / ob имеют распределение α / β-клеток, аналогичное мышам дикого типа, с ожидаемым распределением α-клеток на внешнем краю островков и β-клеток в центре островков, в отличие от более неорганизованная общая архитектура островков об / об .Прямое количественное сравнение площади островков на площадь поджелудочной железы между островками ob / ob и col6KOob / ob выявило в целом более мелкие островки у мышей, лишенных коллагена VI. Таким образом, общий размер и морфология островков у мышей col6KOob / ob показали значительные улучшения.
Мыши col6KOob / ob имеют повышенное количество кавеол плазматической мембраны и измененную передачу сигналов инсулина в жировой ткани. Липидные рафты / кавеолы играют важную роль в передаче сигналов инсулина в адипоцитах посредством стабилизации рецептора инсулина (14).Визуальный осмотр многочисленных адипоцитов с помощью ПЭМ позволяет предположить, что плазматические мембраны адипоцитов col6KOob / ob и col6KO демонстрируют драматические ультраструктурные изменения. Мыши col6KOob / ob имеют резко увеличенное количество кавеол и увеличенное интерстициальное пространство между адипоцитами по сравнению с мышами ob / ob (рис. 3I). В то время как многие из этих кавеолярных инвагинаций плазматической мембраны имеют типичную конформацию в форме колбы, существует значительное количество кавеолярных структур, которые, по-видимому, не прикреплены к плазматической мембране в адипоцитах col6KOob / ob .Напротив, мыши ob / ob имеют более обычное расположение кавеол, что делает очевидным, что изменения в составе внеклеточного матрикса приводят к изменениям инвагинации кавеол с плазматической мембраной. Известно, что кавеолы играют важную роль в функции адипоцитов. Одно хорошо описанное явление — это ассоциация кавеолина-1, ключевого структурного и регуляторного компонента кавеол, с рецептором инсулина. Поэтому мы рассмотрели, изменяет ли измененное состояние кавеолярных структур в адипоцитах col6KOob / ob локальную чувствительность к инсулину в жировой ткани.Как предполагает повышенная плотность кавеол в адипоцитах, мыши col6KOob / ob имеют повышенную активацию AKT при стимуляции инсулином в жировой ткани (Fig. 3J). После 10 мин стимуляции инсулином фосфорилирование AKT в два раза выше у мышей col6KOob / ob , чем у однопометников ob / ob , что указывает на улучшение передачи сигналов инсулина.
Коллаген VI-нулевые мыши имеют увеличенный размер клеток адипоцитов. Улучшение метаболизма обычно связано с уменьшением средней жировой массы и размера клеток.Гистологическое исследование эпидидимальной (рис. 4A) и брыжеечной (данные не показаны) жировой ткани показало, что адипоциты больше в жировой ткани col6KOob / ob по сравнению с адипоцитами их однопометников ob / ob . Это удивительно, поскольку средний размер клеток адипоцитов у мышей ob / ob уже резко увеличен по сравнению с адипоцитами дикого типа. Эта склонность к увеличению размера адипоцитов не является уникальной для мышей col6KO на фоне ob / ob , но также может наблюдаться у мышей col6KO на диете с высоким содержанием жиров (данные не показаны).Количественная оценка распределения адипоцитов по размеру ясно подчеркивает этот сдвиг в сторону больших площадей адипоцитов в модели с нулевым уровнем коллагена VI (рис. 4B). Принимая во внимание общие улучшения метаболизма у мышей col6KOob / ob , это увеличение размера клеток является довольно неожиданным, поскольку улучшения метаболизма обычно связаны с меньшими адипоцитами, а не с большими адипоцитами. Визуализация адипоцитов с помощью сканирующей электронной микроскопии подтвердила больший размер адипоцитов, но также выявила менее ограничивающую среду адипоцитов за счет изменения состава внеклеточного матрикса (рис.4С). Коллаген VI явно представляет собой важный компонент среды внеклеточного матрикса жировой ткани. Недостаток коллагена VI приводит к менее плотной и менее структурированной жировой ткани. Это заставило нас предположить, что это изменение текстуры связано с изменениями общего содержания коллагена в ткани. Два независимых метода были использованы для оценки общего содержания коллагена во внеклеточном матриксе мышей col6KOob / ob . Окрашивание соединительной ткани по Сириусу проводили на жировой ткани, которое не выявило каких-либо серьезных гистологических различий (рис.4D). В качестве более количественной меры оценки также определяли содержание гидроксипролина в ткани. Поскольку гидроксипролины являются основным компонентом коллагенов, количественное определение уровней этой модифицированной аминокислоты считается точным индикатором общего содержания коллагена. Несмотря на менее структурированные, визуально очевидные структурные изменения в жире col6KOob / ob , эти два метода подтвердили, что общее содержание коллагена не изменилось (рис. 4E). Это может быть связано с индукцией других коллагенов в отсутствие коллагена VI.Более вероятно, что это связано с тем, что коллаген VI содержит только относительно незначительные участки классических коллагеновых повторов, которые гидроксипролинируются, и поэтому он не будет иметь значительного влияния на общий гидроксипролин в тканях. Тем не менее, коллаген VI вносит уникальный вклад в стабильность среды внеклеточного матрикса в жировой ткани.
РИС. 4.
Мыши col6KOob / ob имеют более крупные адипоциты. (A) H & E-окрашенные срезы жировой ткани придатка яичка от мышей col6KOob / ob и их однопометников ob / ob .(B) Площадь адипоцитов рассчитывалась из срезов эпидидимальной и брыжеечной жировой ткани с использованием программного обеспечения ImageJ. На графике слева показано процентное содержание адипоцитов в диапазоне площади 2 каждые 100 мкм. На правой панели показаны средние площади адипоцитов в эпидидимальном и брыжеечном жире. Площадь адипоцитов определяли у четырех мышей / группу (подсчитано> 500 клеток / группу). *, P <0,05 по критерию Стьюдента t . (C) Фальшивые сканирующие электронные микрофотографии адипоцитов из жировой ткани придатка яичка.(D) Окрашивание сириусом красного коллагена из эпидидимального жира. (E) Содержание гидроксипролина в эпидидимальном жире было измерено как индикатор общего содержания коллагена. Все эксперименты (от A до E) были выполнены на 8-недельных мышах.
Нас интересовало, можно ли рассматривать состояние ожирения как состояние фиброза и может ли потеря коллагена VI снизить этот уровень фиброза. Таким образом, мы исследовали уровни нескольких генов с четко установленной ролью в фиброзе и обнаружили, что уровни многих ключевых фиброзных генов изменены.Люмикан и декорин представляют собой протеогликаны, которые, как было ранее установлено, связывают фибриллярные коллагены, включая коллаген VI (3, 21, 52), и могут вмешиваться в фибриллогенез коллагена (40, 45). Эти гены принимают активное участие в эпителиально-мезенхимном переходе, который происходит во время фиброза, процесса, во время которого эпителиальные клетки дифференцируются и принимают более мезенхимоподобную морфологию (30, 46). Люмикан оказывает стимулирующее действие на эпителиально-мезенхимальное переходное состояние фиброза, в то время как декорин, напротив, играет ингибирующую роль в трансформации фиброза, индуцированного фактором роста β (TGF-β) (30, 45).Количественная ПЦР в реальном времени показала, что уровни мРНК люмикана снижались, а уровни декорина повышались. Уровни TGF-β также подавлялись в жировой ткани col6KOob / ob (фиг. 5A). При дальнейшем исследовании уровней люмикана и декорина во время различных метаболических состояний мы обнаружили, что уровни люмикана положительно коррелировали с ожирением ( ob / ob и db / db ) и отрицательно с улучшением метаболического состояния (лечение агонистами PPARγ или мыши adipoTG) (Рисунок.5Б). Точно так же TGF-β также регулировался обработкой агонистом PPARγ (фиг. 5C). Это говорит о том, что люмикан и TGF-β играют решающую роль в регуляции фиброза при ожирении. Это может быть подтверждено на уровне белка, где уровень белка TGF-β снижен. Это также приводит к снижению активации нижестоящих сигнальных медиаторов TGF-β, таких как SMAD2 и SMAD3, состояние фосфорилирования которых также снижается (Fig. 5D).
РИС. 5.
У мышей col6KOob / ob снижена передача сигналов TGF-β.(A) Уровни экспрессии генов внеклеточного матрикса (TGF-β1, декорин, люмикан, эластин и несколько MMP) измеряли с помощью qRT-PCR в эпидидимальном жире от 8-недельного ребенка col6KOob / ob и ob / ob мышей. Уровни экспрессии нормализовали до 18S рРНК. (B) Уровни экспрессии люмикана коррелируют с различными метаболическими состояниями. Уровни экспрессии определяли, как описано для фиг. 1D, и представлены как кратное изменение. (C) Уровни TGF-β1 в придатковой жировой ткани 10-недельных мышей, получавших 30 мг / кг COOH или носителя в течение 14 дней.Уровни экспрессии измеряли с помощью qRT-PCR и нормализовали до 18S рРНК. (D) Вестерн-блот-анализ TGF-β и состояния фосфорилирования сигнальных медиаторов TGF-β SMAD2 и SMAD3 в жировой ткани придатка яичка. Интенсивности полос были количественно определены с помощью системы визуализации LI-COR (LI-COR Biosciences) и показаны на нижней панели. Для панелей от A до D данные представлены в виде средних значений ± стандартные ошибки ( n = четыре мыши / группа). *, P <0,05; **, P <0,01 (по критерию Стьюдента t ).
Интересно, что отсутствие коллагена VI также привело к снижению уровней эластина (рис. 5A), ключевой молекулы эластичности матрикса, основная функция которой — возвращать клеткам их исходную форму при растяжении. Это уменьшенное усилие на адипоцитах col6KOob / ob , возвращающееся к своей первоначальной форме, предполагает, что адипоциты способны растягиваться и расширяться с меньшим натяжением со стороны эластиновых волокон, которые обычно пытаются вернуть их в исходное состояние. Огромный спектр матричных металлопротеаз (ММП) также изменяется: MMP-1a и -7 активируются, тогда как MMP-3, -13, -25 подавляются.Ранее было показано, что MMP-3 и -13 положительно коррелируют со степенью ожирения, тогда как MMP-7 отрицательно коррелирует со степенью ожирения (11, 36).
Улучшение метаболизма совпадает с уменьшением воспаления в жировой ткани. Ожирение характеризуется хроническим воспалительным состоянием, во время которого макрофаги рекрутируются в жировую ткань для усиления местной воспалительной программы. Механизмы рекрутирования макрофагов сложны, в процесс вовлечены многочисленные хемокины и цитокины (54, 56).Ожирение также сопровождается увеличением адипоцитов, что увеличивает вероятность некротической гибели непропорционально больших адипоцитов, что может еще больше усилить местное воспаление. Улучшение метаболизма также может обратить вспять этот воспалительный фенотип, связанный с ожирением, и наоборот (28). Поскольку мыши col6KOob / ob показали различные улучшения метаболизма, мы рассмотрели возможные эффекты на воспаление, исследуя инфильтрацию макрофагов у этих мышей.Срезы эпидидимальной жировой ткани окрашивали на F4 / 80, макрофаг-специфический маркер клеточной поверхности. Эти гистологические окраски намекали на уменьшенную инфильтрацию макрофагов у мышей col6KOob / ob по сравнению с мышами ob / ob , хотя это было трудно определить при визуальном осмотре (фиг. 6A). Это было подтверждено более количественной оценкой уровней мРНК F4 / 80 с помощью анализа qRT-PCR, который показал тенденции к снижению уровней этого маркера макрофагов в придатке яичка и к значительному снижению уровней в мезентериальном жире (рис.6Б). Это уменьшение местного воспаления было также подтверждено анализом других воспалительных генов. Уровни мРНК MCP-1 и SAA-3 были значительно снижены в мезентериальном жире, в то время как наблюдалась тенденция к снижению уровней в эпидидимальном жире. Из этого очевидно, что мезентериальный жир более чувствителен к снижению уровней провоспалительных маркеров, чем эпидидимальный жир (рис. 6В). В соответствии с возможной противовоспалительной ролью PPARγ в жировой ткани, не содержащей коллагена VI, уровни мРНК PPARγ повышены в мезентериальном жире у мышей col6KOob / ob (рис.6С).
РИС. 6.
Отсутствие коллагена VI уменьшает системное воспаление и воспаление, специфичное для жировой ткани. (A) Иммуногистохимический анализ F4 / 80 эпидидимальных срезов жировой ткани мышей col6KOob / ob и ob / ob в качестве индикатора степени инфильтрации ткани макрофагами. (B) Количественный анализ ПЦР в реальном времени на воспалительные цитокины F4 / 80, MCP-1 и SAA3 в придаточной и брыжеечной жировой ткани. Уровни экспрессии всех генов были нормализованы до 18S рРНК (среднее значение ± стандартные ошибки; n = четыре мыши / группа).(C) анализ qRT-PCR экспрессии PPAR-γ в мезентериальном жире. Уровни экспрессии были нормализованы до 18S рРНК (среднее значение ± стандартные ошибки; n = четыре мыши / группа). (D) Состояния фосфорилирования ERK, JNK и p38 MAPK в придатковой жировой ткани мышей col6KOob / ob и ob / ob определяли вестерн-блоттингом со специфическими антителами для их фосфорилированных форм и для общих уровней. Интенсивность полос определяли количественно (средние значения ± стандартные ошибки; n = три мыши / группа).(E) Проводили провокацию LPS у мышей с нулевым уровнем коллагена VI и мышей WT. Измеряли уровни циркулирующего IL-6 и MCP-1 (верхняя панель). Уровни экспрессии мРНК воспалительных цитокинов IL-6 и фактора некроза опухоли альфа (TNFα) измеряли в эпидидимальном жире с помощью qRT-PCR и нормализовали до 18S рРНК через 90 минут после инъекции LPS (нижняя панель) (среднее значение ± стандартные ошибки; n = четыре мыши / группа). (F) CLS визуализировали в окрашенных F4 / 80 срезах эпидидимальной жировой ткани. Одиночная звездочка обозначает типичный CLS, а двойная звездочка указывает на остаточную липидную каплю после гибели клетки.(G) Апоптозные адипоциты визуализировали с помощью окрашивания TUNEL срезов эпидидимальной ткани мышей col6KOob / ob и ob / ob . Окрашивание коричневым цветом указывает на TUNEL-положительное ядро (стрелка), а окрашивание в синий цвет (окрашивание гематоксилином) указывает на нереактивное ядро (стрелка). (H) Стресс эндоплазматического ретикулума измеряли с помощью ПЦР-анализа на Xbp-1. Праймеры, охватывающие стык сплайсинга, использовали для амплификации несплайсированных и сплайсированных форм Xbp-1 и разделяли на 3% агарозном геле.Несплицированный Xbp-1 (Xbp1u) составляет 171 п.н., в то время как сплайсированный Xbp-1 (Xbp1s) составляет 145 п.н., и является положительным признаком стресса эндоплазматического ретикулума. Все эксперименты на этом рисунке были выполнены на 10-недельных мышах col6KOob / ob и ob / ob . *, P <0,05 (данные на панелях B, C, D и F были проанализированы с помощью теста Стьюдента t ; данные на панели E были проанализированы с помощью двухфакторного дисперсионного анализа).
Одно из возможных объяснений общего явления, при котором большие адипоциты обычно страдают от воспаления высокой степени, заключается в том, что большая липидная капля внутри адипоцита оказывает значительное давление на плазматическую мембрану в ее попытке дальнейшего расширения.Однако, поскольку адипоциты обычно скованы очень жесткой средой внеклеточного матрикса, напряжение на мембране адипоцитов от расширяющейся липидной капли может вызвать значительную степень напряжения сдвига на мембране. Есть, бесспорно, многие явления, которые могут способствовать увеличению воспаления. Мы обнаружили, что активность ключевых регуляторных киназ, участвующих в стрессе сдвига, ERK и JNK, значительно снижена в адипоците col6KOob / ob . Эти эффекты выражены для ERK и несколько менее драматичны для JNK, в то время как митоген-активируемая протеинкиназа p38 (MAPK) не изменяется.Сниженная активность ERK согласуется со сниженным напряжением сдвига на мембране col6KOob / ob и поддерживает концепцию, что более гибкая среда внеклеточного матрикса может позволить адипоцитам свободно разрастаться без ограничений жесткой внеклеточной среды (рис. 6D).
Коллаген VI-нулевые мыши уменьшили воспаление в ответ на провокацию эндотоксином. Ранее мы показали, что жировая ткань может вносить значительный вклад в системное воспаление, даже в контексте сильных провоспалительных стимулов, таких как эндотоксин.Повышенный базовый уровень воспалительного процесса в жировой ткани, который уже вызван конститутивно повышенной инфильтрацией макрофагов, имеет тенденцию отвечать более высокой амплитудой воспаления на дополнительный системный провоспалительный стимул. Поэтому мы исследовали, трансформируется ли сниженный исходный уровень воспаления у мышей col6KO в снижение ответа на воздействие эндотоксина (рис. 6E). Уровни циркулирующих IL-6 и MCP-1 были фактически ниже у мышей col6KO через 6 часов после заражения LPS (рис.6Е, верхняя панель). Это также отражалось в отношении индукции провоспалительных генов в жировой ткани в этих условиях цитокинами, такими как IL-6 и фактор некроза опухоли альфа, которые были значительно уменьшены после 90 мин воздействия эндотоксина (рис. 6E, нижняя панель).
Адипоциты мышей col6KOob / ob демонстрируют пониженную гибель некротических клеток. Ожирение характеризуется «короноподобными структурами» (CLS), которые состоят из некротических адипоцитов, окруженных активированными макрофагами.Было продемонстрировано, что степень некроза адипоцитов положительно коррелирует с увеличением размера клеток адипоцитов при ожирении (13). Однако, основываясь на иммуногистохимии, профилировании воспалительных генов и сниженной активности MAP-киназы, очевидно, что жировая ткань мышей col6KOob / ob не подчиняется этим общепринятым правилам. Чтобы оценить, приводит ли снижение внеклеточной жесткости, связанной с фенотипом адипоцитов col6KOob / ob к снижению уровня некроза адипоцитов, мы исследовали распределение короноподобных структур в эпидидимальном жире.В соответствии с предполагаемым улучшением выживаемости больших адипоцитов, мыши col6KOob / ob показали пониженное количество CLS (фиг. 6F). Это было подтверждено окрашиванием TUNEL срезов жировой ткани, показавшим пониженную степень гибели клеток в адипоцитах col6KOob / ob (фиг. 6G). Следовательно, вероятно, что более крупные адипоциты у мышей col6KOob / ob способны увеличиваться из-за уменьшения ограничений обычно жесткого внеклеточного матрикса. Это положительно сказывается на их выживаемости, что в дальнейшем приводит к уменьшению воспаления.
Уменьшенные ограничения также очевидны при рассмотрении других маркеров напряжения. Одним из таких маркеров является Xbp1, который является измерителем напряжения в эндоплазматической сети. Он находится в своей основной форме при нормальных условиях, тогда как во время стресса он альтернативно сращивается и мигрирует к ядру. Мыши col6KOob / ob демонстрируют очень значительное снижение сплайсированной формы Xbp1s по сравнению с аналогичными по возрасту одноклетками ob / ob , что отражает снижение уровня клеточного стресса (рис.6H).
Метаболические различия, вызванные отсутствием коллагена VI. Чтобы проверить, может ли отсутствие коллагена VI вызывать какие-либо фундаментальные различия в других метаболических параметрах, мы поместили мышей col6KOob / ob и ob / ob в метаболические камеры. Мы выполнили калориметрические измерения на мышах, которые были старше, в то время, когда больше нет значительных различий в скорости набора веса, абсолютном весе или массе жира между мышами col6KOob / ob и ob / ob .Однако наблюдалась весьма значительная разница в потреблении пищи: мыши col6KOob / ob потребляли только половину того количества пищи, которое потребляли мыши ob / ob , несмотря на сопоставимые массы тела и состав на этой стадии (фиг. 7A). Это можно только частично объяснить небольшим, но значительным снижением передвижения (рис. 7B), особенно в дневное время. Это также отразилось на снижении потребления кислорода (VO 2 ) в течение того же периода времени, что является отражением снижения расхода энергии в дневное время (рис.7C). Кроме того, мыши col6KOob / ob имели измененный коэффициент респираторного обмена (RER), что свидетельствует о различном использовании субстрата. Коэффициент респираторного обмена показывает, окисляются ли преимущественно липиды (RER, 0,7) или углеводы (RER, 1). RER у мышей col6KOob / ob значительно снижается на протяжении всего цикла свет / темнота, указывая на то, что они преимущественно сжигают жирные кислоты, в отличие от мышей ob / ob , которые больше полагаются на углеводы (рис.7D). Это предпочтение использования жирных кислот у мышей col6KOob / ob аналогично стандартным уровням RER у худых мышей, а также является прогнозируемым результатом повышения чувствительности к инсулину, такого как наблюдаемое у мышей col6KOob / ob . В совокупности эти эксперименты показывают очень значительные изменения на уровне потребления пищи и расхода энергии с общей тенденцией к более высокому уровню потребления жирных кислот.
РИС. 7.
Исследования метаболических клеток в клетках на мышах col6KOob / ob и их однопометниках ob / ob .(A) Потребление пищи было измерено и выражено как потребление пищи на грамм веса тела (среднее значение ± стандартные ошибки; n = четыре мыши / группа). (B — D) Общее передвижение (передвижение и выращивание) (B), потребление кислорода (VO 2 ) (C) и RER (D) измерялись в течение дня (средние значения ± стандартные ошибки; n ). = четыре мыши / группа). Возраст всех мышей, использованных в этом эксперименте, составлял 12 недель. *, P <0,05 по критерию Стьюдента t или двустороннему дисперсионному анализу.
ОБСУЖДЕНИЕ
В нормальных условиях жировая ткань считается соединительной тканью с низкой плотностью и высокой пластичностью.Однако здесь мы продемонстрировали, что при прогрессировании ожирения содержание соединительных волокон в жировой ткани резко возрастает из-за активации нескольких коллагенов. По мере увеличения содержания коллагена общая жесткость жировой ткани также увеличивается, что, вероятно, способствует увеличению ее механической прочности. Термин фиброз был определен как образование фиброзной ткани как репаративный или реактивный процесс; он широко используется в отношении печени, легких и почек, а также некоторых других тканей.В жировой ткани фиброз, по-видимому, инициируется в ответ на гипертрофию адипоцитов, которая возникает как начальный шаг к увеличению жировых подушечек за счет увеличения размера липидных капель в существующих адипоцитах. Эта клеточная экспансия, по-видимому, представляет собой начальное «повреждение», в ответ на которое запускается активация компонентов внеклеточного матрикса. В настоящее время мы изучаем, какие клеточные факторы связывают клеточную экспансию адипоцитов с повышенной транскрипционной активностью на уровне коллагенов; Наши предварительные данные позволяют предположить, что фактор 1α, индуцируемый гипоксией, играет в этом процессе решающую роль (N.Halberg et al., Представлены для публикации).
Уровень составляющих внеклеточного матрикса во всех тканях является отражением баланса между скоростью синтеза белков матрикса и их деградацией. Разложение достигается за счет семейства белков, называемых ММП, которые, в свою очередь, регулируются их ингибиторами, называемыми тканевыми ингибиторами ММП. Во время ремоделирования ткани баланс между этими молекулами может измениться, чтобы удовлетворить непосредственные потребности ткани в расширении.Измененная экспрессия протеогликанов люмикан и декорина у мышей col6KOob / ob имеет фундаментальное значение для антифибротической природы жира.
Люмикан участвует в различных процессах, таких как миграция клеток, пролиферация, заживление ран и воспаление. Одна из его наиболее уникальных ролей — его способность связываться и взаимодействовать с макрофагами, взаимодействие, для которого его боковые цепи кератансульфата имеют решающее значение (19). Декорин хорошо известен своей антифибротической ролью в легких, почках, мышцах и печени благодаря своей способности ингибировать TGF-β (25, 27, 30).TGF-β, в свою очередь, играет центральную роль в модулировании баланса между скоростью синтеза и деградации матричных белков. TGF-β способствует фиброзу нескольких тканей: за счет увеличения экспрессии компонентов матрикса, таких как коллагены, фибронектин и протеолгиканы матрикса, и путем индукции ингибиторов разрушающих матрикс ММП. Следовательно, несмотря на то, что люмикан и декорин являются белками, сшивающими коллаген, эти два белка играют противоположную роль в фиброзе. Это может быть связано с их специфической ролью во время фибриллогенеза коллагена или с разнообразными взаимодействиями, которые обеспечивают их уникальные боковые цепи протеогликанов (19, 40).Подавление люмикана и TGF-β и усиление декорина предполагают механизм снижения фиброза жира у мышей ob / ob с дефицитом коллагена VI.
В недавних сообщениях описана роль PPARγ как мощного антифибротического фактора. Агонисты PPARγ могут уменьшать фиброз в нескольких органах, включая почки, печень, сердце и легкие (1, 42, 43, 58). Было высказано предположение, что агонист PPARγ тесаглитазар, член семейства тиазолидиндионов (TZD), снижает гломерулофиброз и отложение коллагена за счет снижения уровня TGF-β1 в почках (8).Сюй и его коллеги продемонстрировали, что PPARγ может действовать как репрессор синтеза коллагена I типа, увеличивая вызванное гамма-интерфероном подавление синтеза коллагена (57). Мы продемонстрировали здесь, что COOH, селективный агонист PPARγ, не основанный на TZD, снижает уровни экспрессии подавляющего большинства коллагенов в жировой ткани. Это обеспечивает альтернативный механизм действия, с помощью которого TZD могут улучшать диабетический фенотип за счет снижения общего содержания внеклеточного матрикса жировой ткани.
Поскольку TGF-β1 играет критическую роль в отложении коллагена, он, вероятно, является мишенью для индуцированного агонистом PPARγ снижения уровня коллагена. Фактически, мы обнаружили, что мыши дикого типа, обработанные агонистом PPARγ, имели пониженные уровни TGF-β1 в жировой ткани (рис. 5C). Снижение уровней TGF-β1 у мышей col6KOob / ob согласуется с возможной ролью TGF-β в индукции фиброза жировой ткани. TGF-β также может активироваться механическим стрессом во многих типах клеток (6, 29, 34, 49, 53).Механическое напряжение на мембране адипоцитов, вызванное расширяющейся липидной каплей, также может быть источником напряжения сдвига на уровне плазматической мембраны. Подобно тому, что сообщалось для многих других тканей в ответ на механический стресс, TGF-β, вероятно, активирует коллагены и другие белки внеклеточного матрикса, чтобы противодействовать этому расширению. Однако во время положительного энергетического баланса адипоцит продолжает расширяться. Повышенная жесткость матрицы жировой ткани противодействует этой тенденции к дальнейшему расширению.Такое перетягивание каната между клеточной экспансией адипоцитов и жесткой внеклеточной средой оказывает огромное давление на плазматическую мембрану, что может привести к гибели клеток из-за некроза. Демеулемейстер и его коллеги продемонстрировали, что у мышей, получавших ингибитор ММР, одновременно с диетой с высоким содержанием жиров, было меньше жировой ткани, увеличилось количество клеток адипоцитов и уменьшился средний диаметр адипоцитов. Их повышенное содержание жирового коллагена в сочетании с уменьшенным размером адипоцитов согласуется с представлением о том, что повышенный коллаген ограничивает рост адипоцитов (17).Пониженный уровень эластина в жировой ткани col6KOob / ob также согласуется с более гибким микроокружением адипоцита.
Одной из основных характеристик ожирения жировой ткани является повышенная частота гибели клеток адипоцитов. Работа Cinti et al. продемонстрировали, что макрофаги собираются вокруг этих мертвых адипоцитов и становятся все более активными в их попытке очистить потенциально цитотоксическую остаточную липидную каплю (13). Эти макрофаги жировой ткани сливаются с образованием многоядерных клеток и окружают каждый мертвый адипоцит короноподобной структурой.В дополнение к этому феномену количество некротических адипоцитов положительно коррелирует со средним размером адипоцитов у мышей с ожирением и других моделей гипертрофии адипоцитов у мышей (13, 50). Мыши с нокаутом по гормон-чувствительной липазе (HSL) демонстрируют гипертрофию адипоцитов и рекрутирование макрофагов без каких-либо других характеристик ожирения. Однако, поскольку HSL-дефицитные мыши обладают типичной жесткой окружающей средой, в отличие от наших мышей col6KOob / ob , вполне вероятно, что адипоциты у HSL-дефицитных мышей сталкиваются с повышенным напряжением сдвига, гибелью клеток и воспалением.
Было показано, что напряжение сдвига и растяжение мембраны активируют членов семейства MAPK в нескольких типах клеток (23, 32, 44, 51). В ответ на напряжение сдвига большие адипоциты демонстрируют повышенный уровень активации пути передачи сигналов β1-интегрина / ERK (18). В некоторых случаях активация JNK, вызванная механическим растяжением, также может приводить к апоптозу клеток (41). Следовательно, напряжение сдвига в больших адипоцитах, по-видимому, является спусковым механизмом для гибели адипоцитов и надвигающегося воспаления.Связь гипертрофии адипоцитов с повышенным рекрутированием макрофагов привела к нынешней догме, согласно которой увеличение размера адипоцитов свидетельствует о воспалении жировой ткани и, как правило, связано с неблагоприятным метаболическим профилем. Напротив, в отсутствие коллагена VI матрица адипоцитов имеет большую гибкость и позволяет адипоцитам продолжать увеличиваться без связанных с ними некроза и воспаления. Это может быть одной из причин, почему мыши col6KOob / ob нарушают установленное в настоящее время правило, согласно которому более крупные адипоциты связаны с усилением воспаления.
Поскольку массив сигнальных молекул, включая множество вышестоящих регуляторов ERK и JNK, находится в кавеолах, было высказано предположение, что активация MAPK, вызванная стрессом сдвига, включает кавеолы (5, 44). Парк и его коллеги продемонстрировали, что воздействие на эндотелиальные клетки напряжения сдвига резко увеличивает количество инвагинированных кавеол. У мышей ob / ob с дефицитом коллагена VI в их адипоцитах наблюдается скопление невагинализированных везикулярных кавеол. Таким образом, различия в кавеолярных структурах мышей col6KOob / ob и ob / ob являются прямым отражением пониженных уровней напряжения сдвига у мышей col6KOob / ob .
Представленные здесь данные согласуются с концепцией, согласно которой уменьшение местного воспаления в жировой ткани мышей col6KOob / ob является основным фактором, способствующим их общему улучшению метаболического фенотипа. Ослабленная среда внеклеточного матрикса в жировой ткани col6KOob / ob позволяет их адипоцитам беспрепятственно разрастаться, тем самым облегчая накопление избыточных липидов и уменьшая эктопическое отложение липидов. Мыши col6KOob / ob также становятся более чувствительными к инсулину, что проявляется в их уменьшении гиперплазии островков и улучшении общей морфологии островков.
Внеклеточный матрикс чрезвычайно важен для структуры и функции практически любого типа клеток; кроме того, он участвует во многих процессах, таких как клеточная адгезия, пролиферация, дифференцировка, миграция, апоптоз и индукция генов (20). В жировой ткани это особенно важно для поддержания структурной целостности адипоцитов и играет решающую роль в адипогенезе. Во внеклеточной среде во время дифференцировки адипоцитов происходят определенные последовательные изменения с каскадом активации и дезактивации различных ММП, дополняющих создание и разрушение коллагеновой сети (31, 33).Обработка преадипоцитов 3T3-L1 ингибитором ММП с широкой специфичностью препятствует образованию полностью дифференцированных липидно-нагруженных адипоцитов (11, 16). Внеклеточный матрикс также претерпевает многочисленные изменения во время ожирения, при этом ММП / тканевые ингибиторы баланса ММП смещаются в сторону увеличения деградации матрикса, предположительно в качестве компенсаторной реакции на повышенное содержание коллагена во время ожирения (11, 17). Несмотря на очевидную значимость матрицы для адипоцитов, очень мало исследований было сосредоточено на тщательной характеристике метаболического воздействия внеклеточного матрикса при ожирении или других метаболических состояниях.
Одним из первых доступных примеров метаболического исследования, посвященного белку внеклеточного матрикса в жировой ткани, было исследование SPARC, матрицеклеточного белка, участвующего во взаимодействиях межклеточного матрикса. SPARC-нулевые мыши имеют большую эпидидимальную жировую подушку и повышенное количество адипоцитов в коже. Несмотря на то, что у нас нет информации о каких-либо метаболических последствиях, которые вызывает отсутствие SPARC, это был первый случай, когда белок внеклеточного матрикса был вовлечен в действие на жировую ткань.Совсем недавно Чун и его коллеги продемонстрировали, что отсутствие матричной металлопротеиназы MT1-MMP вызывает липодистрофию у мышей, нарушая развитие белой жировой ткани из-за прямого воздействия MT1-MMP на дифференцировку адипоцитов (12). Интересно, что эти эффекты можно было наблюдать только in vitro, когда клетки выращивали в трехмерной матрице.
Мы выбрали коллаген VI в качестве кандидата в матричный компонент и изучили его влияние на физиологию жировой ткани. Коллаген VI является идеальным кандидатом из-за его относительно обильной и высокообогащенной экспрессии в жировой ткани.Это наиболее экспрессируемый коллаген в дифференцированных адипоцитах, который претерпевает драматические структурные изменения в процессе адипогенеза (38). Фибриллы коллагена VI изменяют свой внешний вид во время дифференцировки преадипоцитов 3T3-L1, переходя от тонкой текстуры фибрилл на 4-й день дифференцировки к очень толстой в полностью дифференцированных адипоцитах. Несколько исследований дополнительно продемонстрировали, что уровни коллагена VI коррелируют с состояниями гипергликемии и инсулинорезистентности (2, 37). Ранее мы продемонстрировали роль коллагена VI, происходящего из адипоцитов, в прогрессировании роста опухоли молочной железы (26).Во время роста опухоли происходит протеолитическое расщепление, и образующиеся в результате С-концевые фрагменты коллагена VI накапливаются в опухолевых клетках, активируя рецептор NG2 и способствуя митогенному ответу в раковых клетках. Наши данные, описывающие усиление апоптоза эпителия молочных желез в отсутствие белка коллагена VI, совместимы с предыдущими данными, которые продемонстрировали потребность в коллагене VI для предотвращения апоптоза и его стимулирующую роль в пролиферации посредством сигнальных событий AKT (22).Отсутствие коллагена VI приводит к проапоптотическому сигнальному событию, что снижает вероятность пролиферации опухоли.
Наши наблюдения выявили ряд изменений на уровне потребления пищи и расхода энергии: отсутствие триггеров коллагена VI приводит к снижению потребления пищи и снижению расхода энергии с более высоким уровнем потребления жирных кислот. Поскольку эти изменения наблюдаются не только при использовании диеты с высоким содержанием жиров, но и на фоне ob / ob , эти явления не могут быть вызваны исключительно лептином.Неясно, как зарегистрированные изменения в жировой ткани вызывают системные изменения в потреблении пищи и расходе энергии. Это вопросы, представляющие очевидный интерес, которыми мы сейчас занимаемся. Кроме того, мы не можем формально исключить, что коллаген VI оказывает влияние на метаболический контроль посредством действия в тканях, отличных от жира.
Таким образом, наше исследование подчеркивает тот факт, что коллаген VI и, возможно, дополнительные компоненты внеклеточного матрикса чрезвычайно важны в модуляции физиологии адипоцитов.Коллаген VI играет важную роль в фиброзном компоненте ожирения и напрямую влияет на способность адипоцитов расширяться. Участие коллагена VI в ожирении открывает новую главу, в которой сам адипоцит влияет на общую метаболическую функцию; однако теперь очевидно, что среда внеклеточного матрикса, окружающая адипоцит, также может играть важную роль. Потребуются дальнейшие исследования для изучения физиологических последствий фиброза жировой ткани и того, играет ли коллаген VI решающую роль в других фиброзных тканях.
Границы | Метаболические изменения в мононуклеарных клетках периферической крови, изолированных от пациентов с терминальной стадией почечной недостаточности
Введение
Хроническая болезнь почек (ХБП) — это изнурительное состояние, которое может привести к прогрессирующей потере функции почек и назначению пациентов на заместительную почечную терапию (1) . Он определяется как снижение скорости клубочковой фильтрации (СКФ) менее 60 мл / мин / 1,73 м 2 в течение не менее 3 месяцев и классифицируется на пять стадий на основе СКФ (2).Пациенты с ХБП могут прогрессировать до терминальной стадии почечной недостаточности (ТПН) на 5 стадии, когда СКФ составляет менее 15 мл / мин / 1,73 м 2 .
Измененная биоэнергетика митохондрий играет центральную роль в развитии и прогрессировании широкого спектра заболеваний человека, включая заболевания почек (3–5). Клетки генерируют энергию (в форме АТФ), необходимую для клеточного метаболизма, в основном за счет окислительного фосфорилирования в митохондриях (6). Следовательно, измерение функции митохондрий может иметь важное значение для понимания баланса различных энергоемких процессов в физиологии почек.
Анализы крови для определения функции митохондрий — это окончательные и минимально инвазивные тесты, используемые для выявления основных заболеваний. Мы использовали PBMC в качестве модельной системы мониторинга заболеваний для биоэнергетических анализов. PBMC представляют собой гетерогенные клеточные популяции, состоящие из моноцитов, Т-клеток, B-клеток и естественных клеток-киллеров, и являются доступным источником митохондрий и других чувствительных биомаркеров. Интересно, что PBMC обладают способностью отражать системные изменения в организме, а биоэнергетика PBMC недавно была исследована при нескольких хронических заболеваниях (7-10).
В этом исследовании мы использовали популяцию пациентов с ESKD и описали метод, позволяющий измерять потребление кислорода митохондриями в PBMC, выделенных из этой популяции. Мы контролировали митохондриальное дыхание в PBMC с помощью технологии Seahorse XF, которая позволила нам сообщить о количественном анализе окислительного фосфорилирования и гликолиза путем измерения скорости потребления кислорода (OCR) и скорости внеклеточного закисления (ECAR), соответственно. Хорошо известно, что этот метод позволяет высокочувствительным образом оценивать функцию митохондрий с точки зрения нескольких биоэнергетических параметров, включая базальное дыхание, выработку АТФ, утечку протонов, максимальное дыхание, резервную дыхательную емкость и немитохондриальное дыхание (11, 12). ).Затем мы связали биоэнергетические параметры PBMC с изменениями в других элементах митохондриального биогенеза (AMPKα, p-AMPKα, Sirt3, PGC-1α), окислительно-восстановительным состоянием митохондрий и антиоксидантными ответами (SOD2) и морфологией митохондрий (DRP1, OPA1), соответственно. . Наши данные подтверждают мнение о том, что как окислительный, так и гликолитический метаболизм нарушены в PBMC, выделенных от пациентов с ESKD, что может быть результатом вклада различных генетических и патологических факторов, включая дисфункцию механизма цепи транспорта электронов (ETC), ограниченную доступность AMPK. для восстановления внутриклеточных уровней энергии и дисбаланса между процессами деления и слияния митохондрий, что ставит под угрозу способность митохондрий изменять свою морфологию в ответ на изменения окружающей среды.
Материалы и методы
Дизайн исследования
Всего в нашем исследовании приняли участие 50 пациентов (35 пациентов с ESKD на диализе и 15 здоровых). Поскольку для анализа внеклеточного потока требовалось большое количество клеток, мы не включили 11 образцов крови (в том числе 5 контрольных и 6 пациентов) с низким выходом PBMC. Для анализа экспрессии белка мы использовали образцы с достаточным количеством PBMC, оставшихся после биоэнергетического анализа (включая все контроли, но только 11 PBMC пациентов).Оба когортных исследования были проведены в соответствии с принципами, изложенными в Хельсинкской декларации, а протокол исследования был одобрен Наблюдательным советом медицинского центра Университета Раша. Письменное информированное согласие от каждого субъекта было получено до включения.
Выделение PBMC
Человеческие PBMC выделяли из свежей крови (<3 ч после венепункции) центрифугированием в стандартном градиенте плотности. Вкратце, от 7 до 10 мл цельной циркулирующей крови от каждого донора собирали в пробирки BD Vacutainer (Becton Dickinson).Содержимое пробирки разбавляли равным объемом Ca 2+ / Mg 2+ -содержащего фосфатно-солевого буфера (PBS; Gibco) и наслаивали на гиперосмолярный градиент Ficoll-Paque (GE Healthcare) осторожно, не нарушая интерфейс. После центрифугирования при 350 g в течение 30 мин при комнатной температуре с выключенными тормозами плазма в верхнем слое была удалена. Мононуклеарные клетки, расположенные между плазмой и фиколлом, собирали одноразовой пипеткой для переноса и переносили в новую коническую пробирку на 50 мл.Остальную часть пробирки заполняли PBS, и PBMC промывали вращением при 1500 об / мин в течение 10 минут. Супернатант осторожно удаляли и осадок, содержащий PBMC, суспендировали в PBS осторожным пипетированием. Трубка снова заполняется PBS, и этап промывки повторяется еще раз при аналогичных условиях. После последней промывки супернатант сливали пипеткой и осадок ресуспендировали в 0,5–1,0 мл PBS с помощью серологической пипетки на 1 мл. Затем PBMC выделяли с помощью последнего центрифугирования (1000 г при 4 ° C в течение 5 мин) и собирали с помощью среды для замораживания, содержащей среду RPMI-1640 с добавлением 30% FBS и 10% DMSO.В среднем от 5 до 10 миллионов PBMC переносили в криопробирки и хранили при -80 ° C до дня эксперимента.
Подготовка PBMC для биоэнергетического анализа
В день анализа флаконы быстро размораживали и содержимое помещали в 10 мл предварительно нагретой (37 ° C) культуральной среды (RPMI с 10% FBS, 100 Ед / мл пенициллина и 100 ед. / Мл пенициллина и 100 мл). мкг / мл стрептомицина). РВМС центрифугировали при 200 g в течение 5 мин, супернатант удаляли. Клетки ресуспендировали в свежей культуральной среде, подсчитывали и подтверждали жизнеспособность путем исключения трипанового синего (Gibco).Затем собирали PBMC путем вращения 200 г в течение 5 минут и один раз промывали аналитической средой (среда Seahorse XF RPMI, pH 7,4, с 1 мМ пируватом натрия, 2 мМ глутамином и 10 мМ глюкозой). Тем временем мы покрыли 24-луночный культуральный планшет Seahorse тканевым клеем Cell-Tak (Corning) с концентрацией 65 мкг / мл (в растворе, содержащем 0,1 М бикарбоната натрия, pH 8,0 и 1 н. NaOH) на 20 мин. Затем лунки дважды промывали PBS для удаления бикарбоната. После другого центрифугирования при 200 g в течение 5 минут РВМС ресуспендировали в буфере для анализа и загружали в лунки 24-луночного культурального планшета Seahorse по бокам каждой лунки.В частности, мы стремились загрузить 600 000 клеток на лунку (в 150–200 мкл), исходя из нашего опыта с PBMC; однако количество зависит от доступности, и нормализация проводилась на основе содержимого ячеек. Затем планшет центрифугировали при 200 g без тормоза. Состояние адгезии и распространение PBMC на покрытых поверхностях лунок визуально подтверждали под фазово-контрастным микроскопом. Перед переносом планшета на анализ объем доводили до 500 мкл в каждой лунке, медленно и осторожно добавляя дополнительную теплую среду для анализа в каждую лунку.Затем планшет переносили в инкубатор без CO 2 , установленный на 37 ° C, на 45–60 минут до загрузки его в анализатор Seahorse (рис. 1A).
Рис. 1 План эксперимента для анализа внеклеточного потока и типичная кривая OCR с параметрами теста Mito Stress Test. Планшеты с клеточными культурами были покрыты, и PBMC были загружены на планшеты в день эксперимента, тогда как анализатор Seahorse XFe24 был стабилизирован, а сенсорные картриджи были гидратированы днем ранее (A) .Анализатор Seahorse записывает значения OCR (ось y) в зависимости от времени (ось x) до и после инъекций олигомицина, FCCP и ротенона / антимицина A (B) . Используя эти показания OCR, набор Mito Stress Test определяет несколько ключевых параметров функции митохондрий, включая базальное дыхание (BR), АТФ-связанное дыхание (АТФ), запасную (резервную) дыхательную емкость (SRC), утечку протонов (PL), максимальное дыхание. (MaxR) и немитохондриальное дыхание (nMitR) (C) .
Анализ внеклеточного потока
За день до эксперимента анализатор Seahorse XFe24 (Agilent) был включен с помощью программного обеспечения Wave (Agilent), работающего в течение ночи, для стабилизации анализатора.В то же время сенсорный картридж Seahorse XFe24 (Agilent) гидратировали, заполняя каждую лунку 24-луночного рабочего планшета 1 мл калибровочного раствора (Agilent) и погружая в него датчики. Затем стопку вспомогательного планшета и картриджа датчика (FluxPak) поместили в инкубатор без CO 2 , установленный на 37 ° C, на ночь. В день эксперимента FluxPak был извлечен из инкубатора без CO 2 , и порты для инъекций были заполнены следующими соединениями: олигомицин (ингибитор окислительного фосфорилирования), фторкарбонилцианид фенилгидразон (FCCP, митохондриальный разобщитель). ) и ротенон / антимицин А (ингибиторы комплекса I / комплекса III).Эти соединения были предоставлены в виде лиофилизированных порошков в комплекте Mito Stress Test Kit (Agilent), который использовался для исследования функции митохондрий в этом исследовании. Сначала мы приготовили исходные растворы, повторно суспендировав их в среде для анализа, а затем дополнительно разбавили до 10-кратной концентрации перед загрузкой в порты. Мы использовали 1,0 мкМ олигомицина и по 0,5 мкМ ротенона и антимицина А для каждого образца. Однако нам пришлось титровать концентрации FCCP (тестовый диапазон от 1,00 до 2,00 мкМ, с шагом 0,25 мкМ) и выбрать оптимальную концентрацию, обеспечивающую максимальную окислительную способность для каждого образца, поскольку концентрация FCCP варьируется в зависимости от метаболических потребностей организма. ячейки (11).Кроме того, FCCP может быть ингибирующим в высоких концентрациях (13). Объемы загрузки каждого 10-кратного соединения в порты для инъекций составляют 56 мкл олигомицина, 62 мкл FCCP и 69 мкл ротенона / антимицина A. Когда процесс калибровки был завершен (примерно через 20 минут), служебный планшет был заменен культурой. пластина. После короткого (3 мин) периода перемешивания первая серия измерений была записана в базовых условиях. После трех записей в каждую лунку последовательно вводили олигомицин, FCCP и ротенон / антимицин А.Между каждой инъекцией регистрировали по три измерения каждого из OCR и ECAR. Все записи были разделены на 8 минут, а время выполнения стандартного митохондриального стресс-теста составляло около 100 минут.
Расчет метаболических параметров функции митохондрий
Вместе со всеми соединениями и стратегиями инъекций, анализ Mito Stress Test обеспечил измерения базального дыхания, продукции АТФ, максимального дыхания, запаса дыхательной способности, немитохондриального дыхания и утечки протонов.Рисунок 1B демонстрирует типичную кривую OCR и параметры анализа митохондриального стресса. Эти метаболические параметры были рассчитаны для каждой лунки с использованием графика OCR, как показано на Рисунке 1C (12). Затем были рассчитаны средние значения (и планки ошибок) результатов отдельных лунок (n = 4 или 5) для каждого образца (контроля или пациента). Поскольку РВМС подсчитывали непосредственно перед анализом и равное количество РВМС высевали на лунку для каждого образца, данные по морскому коньку были внутренне нормализованы по количеству клеток.
Два дополнительных фактора (например,g., эффективность сцепления и индекс биоэнергетического здоровья) были получены из этих параметров для характеристики биоэнергетики PBMC. Эффективность связывания ETC выражается как отношение (или процент) движущей силы протона, рассеиваемой АТФ-синтазой (то есть отношение оборота АТФ к базальному дыханию). Это мера кислорода, потребляемого для синтеза АТФ, по сравнению с утечкой протонов. Другой параметр, индекс биоэнергетического здоровья (BHI), включает в себя различные биоэнергетические параметры и может быть определен как (14):
BHI = log [Резервная дыхательная способность] a × [производство АТФ] b [Немитохондриальное дыхание] c × [Протон утечка] d (1)
Не применялась степенная функция (т.е.е., экспоненты a, b, c и d были установлены равными 1,0). Он действует как суррогатный индекс изменения биоэнергетического здоровья людей.
Другим измерением метаболической активности является подкисление внеклеточной среды, которое регистрируется как ECAR. Основным фактором подкисления среды является производство лактата во внеклеточной среде. Предполагая, что существует надежная корреляция между ECAR и продукцией лактата, изменения в базальном ECAR в прикрепленных PBMC были использованы для оценки конечной концентрации лактата в клетках, которая отражала их гликолитическую способность.Мы рассчитали базовые скорости подкисления среды (OCR / ECAR при базальном дыхании), чтобы предсказать гликолитические резервные способности PBMC в условиях покоя. Кроме того, резкое увеличение подкисления среды в ответ на олигомицин (OCR / ECAR после добавления олигомицина) и резкое увеличение подкисления среды в ответ на FCCP (OCR / ECAR при максимальном дыхании) также были рассчитаны для оценки гликолитической способности, когда продукция митохондриального АТФ была блокируется олигомицином, и когда искусственная потребность в АТФ вызывается FCCP, соответственно.
Вестерн-блот анализ
Для лизатов целых клеток PBMC ресуспендировали в ледяном буфере для лизиса, содержащем 50 мМ трис-HCl, pH 7,4, 150 мМ NaCl, 1% Nonidet P-40, 0,5% дезоксихолат натрия, 0,1% натрия. додецилсульфат (SDS), 5 мМ EDTA, 1 мМ EGTA (Boston BioProducts) с добавлением протеазы (Roche Diagnostics) и ингибиторов фосфатазы (Sigma Aldrich). После инкубации на льду в течение 30 минут (включая пять этапов импульсного встряхивания) лизаты центрифугировали при 13000 об / мин в течение 15 минут при 4 ° C, чтобы удалить остатки клеток.Лизаты были скорректированы по содержанию белка с использованием анализа Брэдфорда (Bio-Rad) и смешаны с загрузочным буфером для образца LDS и восстанавливающим агентом (оба от Thermo Fisher Scientific) перед денатурированием путем нагревания при 70 ° C в течение 10 минут. Белки разделяли электрофорезом с использованием геля NuPAGE Novex 4–12% Bis-Tris (Thermo Fisher Scientific) и переносили на мембрану Immobilon-P PVDF (Millipore) в соответствии с инструкциями производителя. Для иммуноблоттинга использовали следующие первичные антитела: AMPKα (1: 1000; Cell Signaling Technology, 2532), p-AMPKα (Thr172) (1: 1000; Cell Signaling Technology, 2535), β-актин (1: 1000; Cell Signaling. Technology, 3700), CoxIV (1: 1000; Cell Signaling Technology, 4844), DRP1 (1: 1000; Cell Signaling Technology, 14647), лактатдегидрогеназа (1: 1000; Cell Signaling Technology, 2012), OPA1 (1: 1000 ; Cell Signaling Technology, 80471), PGC-1α (1: 1000; Cell Signaling Technology, 2178), PDHK1 (1: 1000; Cell Signaling Technology, 3820), Sirt3 (1: 1000; Cell Signaling Technology, 2627) и SOD2 (1: 1000; Cell Signaling Technology, 13141).После инкубации с первичными антителами в течение ночи при 4 ° C мембраны зондировали вторичными антителами, конъюгированными с HRP (оба 1:10 000) против кроличьих (Promega, W401B) или против мышиных (Promega, W402B), в течение 1 часа. Белки детектировали с помощью реагента ECL (Thermo Fisher Scientific), а авторадиографические пленки (Denville) проявляли с использованием процессора рентгеновских пленок (Alphatek). Интенсивность сигналов измеряли денситометрией с использованием программного обеспечения Image J (NIH).
Статистический анализ
Эксперименты по анализу функции митохондрий повторяли в четырех или пяти лунках, тогда как анализы экспрессии белка независимо повторяли два или три раза.Результаты были усреднены для каждой группы лечения. Данные представлены как средние значения ± стандартная ошибка средних значений (SEM). T-критерий Стьюдента использовался для оценки различий в экспериментальных характеристиках с помощью программного обеспечения Prism 6 (GraphPad). Значимые различия по сравнению с контролем (CTL) аннотированы следующим образом: * P <0,05, ** P <0,01 и *** P <0,001.
Результаты
Характеристики исследования и участников
Всего в 29 образцах пациентов и 10 контрольных образцах было достаточно PBMC для метаболического анализа (таблица 1).Девяносто процентов пациентов страдали гипертонией и 62% страдали диабетом.
Таблица 1 Основные демографические и клинические характеристики исследуемой группы (АГ, гипертония; СД, сахарный диабет; Ж, женщина; М, мужчина; АА, афро-американец; CAU, европеоид; HSP, латиноамериканец).
Уровни потребления кислорода и биоэнергетические параметры контрольной группы и пациентов с ESKD
Чтобы изучить уровни потребления кислорода и биоэнергетические параметры контрольной группы и пациентов с ESKD, мы использовали анализатор Seahorse XFe24 в живой клеточной среде (рис. 2A).
Рис. 2 Митохондриальные профили PBMC, выделенных от пациентов с ESKD и здоровых контролей. Профили потребления кислорода пациентами PBMC (заболевание, n = 29) и здоровыми пациентами (здоровье, n = 10), зарегистрированные до и после добавления олигомицина, FCCP и ротенона / антимицина A, соответственно (A) . Анализ сообщает о значительно более высоком базальном дыхании (B) , АТФ-связанном дыхании (C) , максимальном дыхании (E) , резервной дыхательной способности (F) и немитохондриальном дыхании (G) в PBMC здоровых субъектов по сравнению с PBMC пациентов с ESKD.Обе группы имеют одинаковые уровни утечки протонов (D) . Эффективность сцепления (H) и индекс биоэнергетического здоровья (I) также оценивались с использованием этих параметров. Круговые диаграммы показывают процентный вклад каждого из АТФ-связанного дыхания (АТФ), утечки протонов (PL), резервной дыхательной емкости (SRC) и немитохондриального дыхания (nMitR), когда максимальный OCR установлен как 100% для здоровья и болезни (Дж) . ** P <0,01 и *** P <0,001.
Сначала мы установили OCR «в состоянии покоя» (OCR начальный ), который учитывает базальную скорость дыхания в PBMC после удаления вклада немитохондриального потребления кислорода (рис. 1B).Мы обнаружили, что PBMC, выделенные от здоровых людей, имели значительно более высокое базальное дыхание по сравнению с пациентами с ESKD (31,9 ± 3,1 против 16,0 ± 1,0 пмоль / мин / 10 5 клеток, P <0,001), что указывает на гораздо более высокий уровень эндогенного АТФ. потребность в контрольных ячейках (рис. 2В).
Затем PBMC обрабатывали олигомицином, который снижает поток электронов через ETC за счет ингибирования АТФ-синтазы. Результирующее снижение митохондриального дыхания после инъекции олигомицина (OCR oligo ) изолирует OCR, связанный с выработкой АТФ (рис. 1B).Потребление кислорода, которое используется для выработки митохондриального АТФ, было выше в контрольных PBMC, чем в клетках, выделенных от пациентов (29,6 ± 2,8 против 14,1 ± 0,9 пмоль / мин / 10 5 клеток, P <0,001, рис. 2C), что отражает пониженную способность PBMC пациентов использовать мембранный потенциал, который служит движущей силой для синтеза АТФ (15). Мы также измерили диссипацию энергии при утечке протонов (рис. 1В) и обнаружили, что обе группы продемонстрировали схожее дыхание, связанное с утечкой протонов (2,3 ± 0,6 и 1,9 ± 0.2 пмоль / мин / 10 5 клеток в состоянии здоровья и болезни соответственно), как показано на рисунке 2D.
Последующая обработка митохондриальным разобщителем, FCCP, перемещает протоны через внутреннюю мембрану, заставляя ETC работать с максимальной мощностью (16). Результатом было максимальное считывание OCR (OCR FCCP ), поддерживающее движущую силу протона на самом высоком уровне (Рисунок 1B). PBMC от здоровых людей смогли достичь более высоких уровней окислительного дыхания после лечения FCCP, чем от пациентов (141.6 ± 17,9 по сравнению с 60,8 ± 5,8 пмоль / мин / 10 5 клеток, P <0,01, рисунок 2E). Получение кривой максимального дыхания также позволило нам рассчитать запасную (или резервную) дыхательную способность, то есть разницу между стимулированным FCCP и OCR в состоянии покоя (рис. 1B). PBMC, полученные от здоровых контролей, имели значительно более высокую резервную респираторную способность, чем у пациентов (109,7 ± 16,7 против 44,8 ± 5,0 пмоль / мин / 10 5 клеток, P <0,01, фигура 2F).
На заключительном этапе митохондриального стресс-теста ротенон и антимицин А были добавлены для блокирования комплексов I и III, соответственно.Вместе эти агенты полностью ингибируют перенос электронов в митохондриях, и измеренный остаточный OCR можно отнести к немитохондриальным оксидазам (ротенон OCR / AA ). Это остаточное дыхание было почти в три раза выше в МКПК, выделенных из контроля, чем у пациентов (14,8 ± 16,7 против 4,9 ± 5,0 пмоль / мин / 10 5 клеток, P <0,001, рис. 2G), что указывает на то, что немитохондриальное дыхание вносит свой вклад. к общему дыханию в большей степени в контрольных PBMC, чем в PBMC пациентов.
Мы обнаружили, что эффективность связывания ниже в PBMC от пациентов с ESKD (0,93 ± 0,01 против 0,88 ± 0,01, P <0,01, рис. 2H), предполагая, что митохондриальная эффективность окисления субстрата ниже в этих клетках по сравнению со здоровым контролем. С другой стороны, BHI не отличался достоверно в обеих группах (2,1 ± 0,2 против 1,9 ± 0,1, рис. 2I). Это открытие удивительно, поскольку мы ожидали, что здоровье митохондрий было нарушено в PBMC пациентов, учитывая относительно низкую производительность этих клеток с точки зрения различных биоэнергетических параметров (Рисунки 2B, C, E – H).Это можно объяснить тем фактом, что каждый параметр в этом уравнении одинаково влияет на общее дыхание в обеих группах, то есть утечка протонов обеспечивает от 1,5% (контроль) до 2,9% (пациенты), немитохондриальное дыхание - 7,5% (пациенты). до 9,5% (контрольная группа), производство АТФ обеспечивает от 18,9% (контрольная группа) до 21,5% (пациенты), а резервная респираторная способность обеспечивает от 68,2% (пациенты) до 70,1% (контрольная группа) от общих служб респираторной способности (Рисунок 2J). В противном случае нашу оценку BHI (уравнение 1) необходимо модифицировать с помощью обновленных показателей, чтобы прогнозировать состояние здоровья всесторонним образом.
Внеклеточное подкисление с помощью PBMC
Мы также смогли обнаружить гликолитическую функцию PBMC, измерив скорость внеклеточного подкисления (ECAR) в наших клетках, которая показывает уровни молочной кислоты, генерируемой гликолизом (рис. 3A). Кривая ECAR была похожа на кривую OCR, за исключением того, что добавление олигомицина вызывало потерю OCR (рис. 2A), но компенсаторное увеличение ECAR (рис. 3A), т.е. гликолиз усиливается в ответ на ингибирование потребления кислорода митохондриями.Сначала мы оценили изменение внеклеточного закисления (ΔECAR) в ответ на олигомицин (рис. 3B), используя общий график ECAR (рис. 3A). PBMC от здоровых субъектов имели более устойчивый ответ на олигомицин (3,2 ± 0,3 против 2,1 ± 0,2, P <0,01), указывая на то, что, по сравнению с PBMC от пациентов с ESKD, эти клетки более легко переключались на гликолиз, когда АТФ-синтаза (и митохондриальное дыхание) было подавлено.
Рис. 3 Скорость внеклеточного закисления в PBMC из-за продукции лактата и CO 2 посредством гликолиза и митохондриального дыхания, соответственно.Внеклеточное закисление, как измерено, выше в PBMC здоровых субъектов на протяжении всего анализа (A) , что дает более сильный ответ после олигомицина в этой группе (B) . Соответствующие соотношения OCR: ECAR в PBMC при базальном дыхании (C) , после лечения олигомицином (D) и при максимальном дыхании (E) также были выше у здоровых субъектов, чем у пациентов с ESKD. * P <0,05, ** P <0,01 и *** P <0.001.
Затем мы проанализировали отношения OCR / ECAR, чтобы оценить относительный вклад гликолиза и окислительного фосфорилирования в выработку энергии (рисунки 3C – E). Мы обнаружили более высокие соотношения OCR / ECAR на исходном уровне (7,2 ± 0,8 против 4,8 ± 0,4, P <0,05, рис. 3C) после блокады АТФ-синтазы (1,8 ± 0,2 против 1,1 ± 0,1, P <0,01, рис. 3D). ) и условий максимального дыхания (9,9 ± 0,6 против 6,4 ± 0,6, P <0,001, рис. 3E), что согласуется с биоэнергетическими профилями обеих групп (рис. 2A и 3A).
Интеграция метаболического анализа с вестерн-блоттингом
Мы измерили уровни экспрессии подмножества митохондриальных белков и других белков, которые влияют на энергетический метаболизм клеток (рис. 4). AMP-активированная протеинкиназа (AMPK) является одним из этих ключевых регуляторов энергетического гомеостаза (17, 18). Мы обнаружили, что экспрессия и активация AMPKα (т.е. фосфорилирование AMPKα на Thr172) были значительно выше в контрольных PBMC по сравнению с PBMC пациентов (P <0,05 для обоих, рисунок 4A).Сиртуины представляют собой еще одну важную группу сенсоров энергии (19), а сиртуин 3 (Sirt3) является новым регулятором митохондриальной биоэнергетики и генерации АТФ (20). Было показано, что активация Sirt3 увеличивает митохондриальное дыхание (21, 22), но мы обнаружили сопоставимые уровни экспрессии этого белка митохондриального матрикса в обеих группах (рис. 4A). Затем мы протестировали уровни коактиватора-1α (PGC-1α) рецептора, активируемого пролифератором пероксисом (PPARγ), поскольку он является важным фактором транскрипции, регулирующим митохондриальный окислительный метаболизм и расход энергии (23, 24).Вестерн-блот-анализ антитела PGC-1α выявил более высокую экспрессию в PBMC от здоровых субъектов (фиг. 4A).
Рисунок 4 Функциональные единицы клеточного метаболизма, регулирующие биоэнергетический аппарат. (A) Репрезентативные вестерн-блоты лизатов PBMC (слева) от пациентов с ESKD (n = 11) и здоровых контролей (n = 10) для ключевых белков, которые участвуют в биоэнергетике и митохондриальной функции (AMPKα, p-AMPKα , Sirt3, PGC-1α, LDH, PDHK1 и SOD2) и соответствующие денситограммы (справа). (B) Репрезентативные вестерн-блоты белков деления митохондрий (DRP1) и слияния (OPA1), а также контрольных антител (вверху) и интенсивности их полос по отношению к домашним белкам β-актина (внизу, слева) и CoxIV (Нижний правый). * Р <0,05.
Затем мы сосредоточились на пируватном узле, который связывает гликолиз с циклом трикарбоновой кислоты (TCA). Мы искали субстратные катаболические пути, питающие цикл TCA, чтобы выяснить, связаны ли низкие скорости дыхания в PBMC от пациентов с ESKD: i) направлением продуктов гликолиза на лактат, а не циклом TCA, или ii) блокадой катализа пирувата до ацетил-CoA. , который входит в цикл TCA для последующих зависимых от кислорода процессов производства энергии.Первый катализируется лактатдегидрогеназой (ЛДГ) (25), тогда как второй катализируется пируватдегидрогеназой (ПДГ), но подавляется киназами пируватдегидрогеназы (ПДГК) (26). Мы обнаружили сопоставимые уровни каждого из LDH и PDHK1 в обеих группах (рис. 4A), предполагая, что резкие различия в окислительном фосфорилировании между контрольной группой и группами пациентов не были связаны с контролем над этим метаболическим переключением вокруг пирувата.
Затем мы хотели изучить механизмы, связанные с окислительным стрессом, путем оценки антиоксидантного статуса наших PBMC.Мы проанализировали экспрессию митохондриально-специфичного фермента, улавливающего свободные радикалы, супероксиддисмутазы 2 (SOD2), и обнаружили более высокую экспрессию в PBMC от пациентов с ESKD (рис. 4A), что указывает на усиление системы антиоксидантной защиты, которая, вероятно, коррелировала с более высокий уровень окислительного повреждения этих клеток (27).
Чтобы исследовать связь между функцией митохондрий и морфологией, мы исследовали уровни DRP1 и OPA1, которые участвуют в делении и слиянии митохондрий, соответственно.Оба митохондриально-образующих белка экспрессировались меньше в PBMC пациентов, чем у здоровых субъектов, когда уровни экспрессии были нормализованы для внутренних эталонных антител β-актина (цитоскелет) или CoxIV (митохондриальный) (рисунок 4B), что указывает на дисфункцию в динамике митохондрий.
Обсуждение
В этом исследовании мы определили уровни потребления кислорода в PBMC, выделенных от пациентов с ESKD, которые связаны с митохондриальным дыханием. Мы обнаружили, что базальное, АТФ-связанное и максимальное потребление кислорода в группе пациентов было ниже, чем в контрольных PBMC, что указывает на нарушение функции митохондрий, что приводит к снижению компонентов механизма ETC и снижению окислительного фосфорилирования при заболевании почек. .Несмотря на то, что окислительное фосфорилирование тесно связано с переносом электронов в нормальных условиях, энергия, производимая во время окисления субстрата, не полностью связана с синтезом АТФ. Некоторые протоны, которые перекачиваются из митохондриального матрикса в митохондриальное межмембранное пространство, чтобы войти в ETC, просачиваются обратно через внутреннюю мембрану митохондрий через разобщающие белки (28). Уровень утечки протонов был сопоставим по здоровью и болезням, что подрывало его влияние на работу митохондрий.Другим важным параметром была резервная респираторная способность, которая показывает, насколько хорошо клетки оснащены для увеличения своей способности к синтезу митохондриального АТФ (29), и измеряется по изменению окислительного фосфорилирования после разобщающей стимуляции. Контрольные PBMC обладают лучшей способностью поддерживать резерв мощности по выработке энергии и реагировать на неожиданное увеличение потребности в энергии (например, при остром поражении, таком как резкие изменения в активации ионного насоса). PBMC, изолированные от пациентов с ESKD, не могли достичь столь высоких уровней окислительного фосфорилирования, которые достигаются контрольными PBMC после стимуляции разобщением, что может быть связано с уменьшенной массой или нарушенной целостностью митохондрий (30).Соответственно, эффективность связывания была ниже в PBMC пациентов в основном из-за дыхания, связанного с альтернативными путями протонной проводимости в этих клетках, что обычно приводит к выработке тепла за счет синтеза АТФ (31).
Мы также измерили скорость внеклеточного закисления, которая приписывается гликолизу. Фактически, внеклеточное закисление вызывают различные пути, помимо гликолиза, такие как бикарбонат (32) и углекислый газ, получаемый из дыхания (CO 2 ) (33).Чтобы исключить его вклад в подкисление, бикарбонат был удален из среды для анализа; в противном случае он будет связываться с протонами, образующимися в результате гликолиза, и диссоциировать с образованием воды (H 2 O) и CO 2 , которые в конечном итоге дегазируют и вызывают изменения pH. Корреляция вклада CO 2 была более сложной задачей, поскольку требовалось выполнить стресс-тест митохондрий с использованием среды для анализа, не содержащей глюкозы. В этом случае подкисление будет происходить в основном из-за гидратации и диссоциации CO 2 , а не за счет гликолиза, который будет демонстрировать ECAR-модель, отражающую только вклад CO 2 .Из-за ограниченной доступности контрольных образцов и образцов пациентов мы не использовали эту стратегию для вычитания вклада CO 2 , продуцируемого во время митохондриальной активности (т. Е. Выработки энергии в цикле TCA), но предположили, что концентрация лактата в PBMC отражает их гликолитическую способность. (34). Следовательно, мы можем получить немного увеличенные значения ECAR, поскольку CO 2 вносит существенный вклад во внеклеточное закисление. В результате анализа были получены кривые ECAR, аналогичные графикам OCR, т.е.е., гликолиз в контрольных клетках регулировался в большей степени по сравнению с пациентами, когда потребление кислорода митохондриями подавлялось. Наши результаты свидетельствуют о том, что энергия преимущественно генерируется окислительным фосфорилированием в PBMC от здоровых субъектов, а PBMC от пациентов с ESKD были относительно более зависимы от гликолиза по сравнению с контрольными PBMC. Однако эти результаты не исключают ни существования устойчивой гликолитической активности в контрольных PBMC, ни остаточного уровня окислительного фосфорилирования в PBMC пациентов.
Дальнейший анализ путей, обеспечивающих цикл TCA в пируватном узле (то есть на последних стадиях гликолиза), показал, что уровни экспрессии каждого из LDH, который восстанавливает пируват до лактата, и PDHK1, который подавляет комплекс пируватдегидрогеназы и ингибирует превращение пирувата в ацетил-КоА, были аналогичными у здоровых субъектов и пациентов с ESKD. Первый механизм позволяет последующую экскрецию лактата во внеклеточное пространство, тогда как ацетил-КоА передает источники углерода в цикл TCA для производства энергии (таким образом, активность обоих ферментов служит для перенаправления пирувата на производство лактата, а не цикла TCA).Эти результаты указывают на то, что плохое дыхание в PBMC пациентов может быть связано не с ферментами или метаболитами, связанными с пируватным узлом, а с производством самого пирувата, поскольку анализ гликолитической функции в PBMC пациентов показал, что этот путь также работает в скорость ниже, чем у элементов управления. Одно небольшое замечание: даже несмотря на то, что скорость гликолиза и продукции пирувата была оценена с помощью скорости внеклеточного закисления [ECAR (34)], в пути гликолиза есть реакции, которые способствуют подкислению и не связаны с производством лактата ( 35).
Одним из путей, который выделяется как значимый для PBMC пациентов, является путь AMPK. AMPK является серин / треониновой киназой и, как известно, опосредует несколько сигнальных путей для поддержания функции митохондрий. Он активируется, когда соотношение АМФ: АТФ увеличивается в результате клеточного ответа на стресс, и как только это происходит, он играет ключевую роль в компенсации пониженного уровня АТФ (36). Другими словами, он определяет истощение АТФ, а затем включает реакции, производящие АТФ, при выключении реакций, потребляющих АТФ.Он регулирует различные метаболические процессы и непосредственно участвует или не регулируется при основных хронических заболеваниях (37, 38). И AMPK, и его активированная форма (через фосфорилирование по остатку треонина, Thr-172, α-субъединицы) были экспрессированы на значительно более низких уровнях в PBMC от пациентов с ESKD, что подчеркивает его важную роль в поддержании гомеостаза клеточной энергии у этих пациентов. У здоровых субъектов мы также обнаружили высокие уровни PGC-1α, что является разумным, поскольку измеренная скорость окислительного фосфорилирования была выше в контрольных PBMC по сравнению с PBMC пациентов, а высокие уровни PGC-1α были связаны с увеличением биоэнергетических требований (39). .Интересно, что исследования на животных продемонстрировали, что фибробласты, полученные от мышей, нулевых по PGC-1α, обладают большей чувствительностью к окислительному стрессу по сравнению с фибробластами дикого типа (40), что может объяснять значительно нарушенное митохондриальное дыхание в PBMC пациентов. Это открытие также отражало результаты блоттинга AMPKα, поскольку AMPK, как сообщалось, активирует PGC-1α как in vivo, , так и in vitro (41). Более того, было обнаружено, что уровни мРНК нескольких нижележащих генов-мишеней PGC-1α значительно подавляются в PBMC, выделенных от диализных пациентов (42).
Новые данные исследований свидетельствуют о том, что нарушение митохондриального дыхания и активности ETC приводит к образованию свободных радикалов, что в конечном итоге способствует усилению окислительного стресса (43). На следующем этапе мы хотели посмотреть, как PBMC реагируют на окислительный стресс, оценивая уровни экспрессии важного митохондриального антиоксидантного фермента SOD2, который катализирует превращение супероксида в пероксид водорода и молекулярный кислород путем связывания с супероксидными побочными продуктами окислительного фосфорилирования ( 44, 45).Мы обнаружили более высокую экспрессию SOD2 в PBMC пациентов, что согласуется с более ранними исследованиями, в которых сообщалось о более высокой экспрессии гена SOD2 у пациентов, находящихся на гемодиализе (46) и перитонеальном диализе (42), по сравнению со здоровыми людьми из контрольной группы. В других исследованиях также сообщалось об увеличении количества активных форм кислорода (АФК) в митохондриях у пациентов с ХБП (47) или ESKD (48) и повышении регуляции антиоксидантных регуляторных генов в PBMC у пациентов с CKD или ESKD (49). В подтверждение этих результатов AMPKα-дефицитные эмбриональные фибробласты мыши показали на 50% больше митохондриальных ROS, что указывает на то, что AMPK действует как сенсор продукции митохондриальных ROS и последующей экспрессии антиоксидантных генов (50).
Митохондрии — это высокодинамичные органеллы, которые постоянно меняют свою форму и число от до хорошо охарактеризованных механизмов, деления и слияния, в основном для восстановления, адаптации или удовлетворения потребностей в энергии (51). Митохондриальная динамика также рассматривается как важный процесс, способствующий митохондриальной дисфункции (52, 53). Следовательно, мы хотели увидеть уровни экспрессии двух основных белков аппарата митохондриального морфогенеза, DRP1 (54, 55) и OPA1 (56, 57), которые участвуют в делении и слиянии митохондрий, соответственно.Мы обнаружили, что оба белка были менее распространены в PBMC у пациентов с ESKD по сравнению с таковым у здоровых субъектов, что указывает на неравновесие между относительными скоростями деления и слияния. Это может изменить ультраструктурную морфологию митохондрий и потенциально нарушить функцию митохондрий в PBMCs, выделенных от пациентов с ESKD. Есть также доказательства того, что AMPK может регулировать механизм митохондриального деления и слияния, опосредуя эти события (58), что может объяснить снижение уровней DRP1 и OPA1 в клетках ESKD, демонстрирующих низкую активность AMPK.
Ограничением настоящего исследования является то, что наши контрольные субъекты были моложе пациентов. Генетические различия также могут иметь место, поскольку расовый состав варьировался среди населения каждой группы (таблица 1). Эти различия в возрасте и расе необходимо учитывать при интерпретации наших выводов. Поскольку PBMC представляют собой гетерогенную популяцию иммунных клеток, и каждая субпопуляция может иметь уникальный биоэнергетический профиль (59), невозможность определить долю каждого типа клеток в здоровых субъектах и образцах ESKD является слабым местом этого исследования.С другой стороны, PBMC могут отражать общее состояние метаболизма и обеспечивать идеальную модель для мониторинга системных изменений (60). В последние годы характеристики потребления кислорода PBMC используются в качестве показателя метаболической функции при широком спектре заболеваний, таких как СД 2 типа (7), синдром хронической усталости (9), инфекция, вызванная вирусом иммунодефицита человека (ВИЧ) (10). , Болезнь Альцгеймера (61) и сепсис (62). Кроме того, размер выборки был относительно скромным. Более того, мы не смогли протестировать все образцы с помощью сомнительного вестерн-блоттинга из-за ограниченного объема образца, а количество образцов, используемых для этого анализа, варьировалось в зависимости от наличия PBMC в каждой из здоровых и больных групп.Было бы интересно проверить эти результаты на более широкой популяции. Проблемы с набором пациентов также повлияли на дизайн нашего исследования: мы собирали образцы крови и изолированные PBMC в день посещения пациента, но позже нам пришлось заморозить PBMC для биоэнергетического анализа. Несмотря на то, что в исследованиях, изучающих функцию митохондрий в клетках крови, обычно используются свежевыделенные клетки, криоконсервация может использоваться при разработке различных исследований, включая лейкоциты (63), лимфоциты (12) и PBMC (9).Также доступны отчеты об оценке качества криоконсервации, в которых предполагается, что замороженные PBMC, если они должным образом криоконсервированы, являются привлекательной альтернативой свежевыделенным клеткам для функциональных исследований (64, 65). Действительно, эти ограничения являются предметом дальнейших улучшений, но могут быть уравновешены применимостью и эффективностью нашей методологии.
Взятые вместе, эти эксперименты с PBMC показывают, что у пациентов с ESKD постоянно сообщалось о митохондриальных нарушениях, и сильно сниженные биоэнергетические параметры могут быть особенностью этих пациентов.Кроме того, мы демонстрируем, что ESKD снижает активность AMPKα, а также уровни экспрессии других белков и факторов транскрипции, регулирующих митохондриальный биогенез, но увеличивает экспрессию антиоксидантной SOD2 как защитного механизма против митохондриальных ROS, которая, вероятно, повышена из-за нарушение оксидативного гемостаза при ХПН. Наши данные также предполагают аномальную митохондриальную динамику при заболевании, о чем свидетельствуют низкие уровни экспрессии белков деления (DRP1) и слияния (OPA1).Более ранние исследования также связывали дисфункцию митохондрий с этиологией заболеваний в контексте заболеваний почек (42, 66), но метаболический генотип клеток (например, биоэнергетические параметры, сдвиг между дыханием и гликолизом и т. Д.) Был более подробно охарактеризован в наше исследование. В целом, наш протокол и данные свидетельствуют о том, что метаболизм митохондрий в почках может быть связан с биоэнергетическими профилями клеток крови.
Заключение
Мы описали методологию оценки фенотипов митохондриального дыхания путем включения инструментов и стандартизации протоколов внеклеточного потока.Наша стратегия показала многообещающие результаты с точки зрения измерения функции митохондрий у пациентов (30, 67) и позволила нам сравнить митохондриальную биоэнергетику в состоянии здоровья и болезни. Учет различий в биоэнергетических возможностях пациентов с ESKD и здоровых лиц из контрольной группы важен, потому что такая информация дает больше информации о том, почему определенные люди подвергаются высокому риску развития и прогрессирования почечной недостаточности. Это улучшит наше понимание механизма заболевания и поможет нам определить новые диагностические цели и разработать более безопасные методы лечения пациентов с ESKD, находящихся на диализе.
Заявление о доступности данных
Исходные материалы, представленные в исследовании, включены в статью / дополнительные материалы. Дальнейшие запросы можно направить соответствующему автору.
Заявление об этике
Исследования с участием людей были рассмотрены и одобрены институциональным наблюдательным советом Медицинского центра Университета Раша. Пациенты / участники предоставили письменное информированное согласие на участие в этом исследовании.
Вклад авторов
MA, SD, LT, BS и HW разработали и разработали исследования.МА, СД и БС проводили эксперименты. MA и SD проанализировали данные и составили рукопись. HW отредактировал рукопись. Все авторы предоставили комментарии к рукописи и одобрили представленную версию. Все авторы внесли свой вклад в статью и одобрили представленную версию.
Конфликт интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.
Благодарности
Авторы благодарят всех, кто участвовал в исследовании, сдав образцы крови.
Ссылки
2. Левей А.С., Корэш Дж., Балк Э., Кауш А.Т., Левин А., Стеффес М.В. и др. Национальный фонд почек. Практические рекомендации Национального фонда почек при хронической болезни почек: оценка, классификация и стратификация. Ann Intern Med (2003) 139: 137–47. doi: 10.7326 / 0003-4819-139-2-200307150-00013
PubMed Аннотация | CrossRef Полный текст | Google Scholar
4. Эмма Ф., Монтини Дж., Парих С.М., Сальвиати Л. Митохондриальная дисфункция при наследственном заболевании почек и остром повреждении почек. Нат Рев Нефрол (2016) 12: 267–80. doi: 10.1038 / nrneph.2015.214
PubMed Аннотация | CrossRef Полный текст | Google Scholar
5. Хайек С.С., Лиф Д., Тахан А.С., Раад М., Шарма С., Вайкар С.С. и др. SuPAR как патогенный фактор и терапевтическая мишень при остром повреждении почек. N Engl J Med (2020) 382: 416–26. doi: 10.1056 / NEJMoa1
1
PubMed Аннотация | CrossRef Полный текст | Google Scholar
7. Хартман М.Л., Ширихай О.С., Холбрук М., Сюй Г., Кочерла М., Шах А. и др.Связь потребления кислорода митохондриями мононуклеарными клетками периферической крови с функцией сосудов при сахарном диабете 2 типа. Vasc Med (2014) 19: 67–74. doi: 10.1177 / 1358863X14521315
PubMed Аннотация | CrossRef Полный текст | Google Scholar
8. Калтон Е.К., Кин К.Н., Райзел Р., Роулендс Дж., Соарес М.Дж., Ньюсхолм П. Изменение статуса витамина D с зимы на лето снижает системное воспаление и биоэнергетическую активность мононуклеарных клеток периферической крови человека. Редокс Биол (2017) 12: 814–20.doi: 10.1016 / j.redox.2017.04.009
PubMed Аннотация | CrossRef Полный текст | Google Scholar
9. Tomas C, Brown A, Strassheim V, Elson JL, Newton J, Manning P. У пациентов с синдромом хронической усталости нарушается клеточная биоэнергетика. PloS One (2017) 12: e0186802. doi: 10.1371 / journal.pone.0186802
PubMed Аннотация | CrossRef Полный текст | Google Scholar
10. Gangcuangco LMA, Mitchell BI, Siriwardhana C, Kohorn LB, Chew GM, Bowler S, et al.Окислительное фосфорилирование митохондрий в мононуклеарных клетках периферической крови снижено при хроническом ВИЧ и коррелирует с иммунной дисрегуляцией. PloS One (2020) 15: e0231761. doi: 10.1371 / journal.pone.0231761
PubMed Аннотация | CrossRef Полный текст | Google Scholar
11. Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. Анализ и интерпретация данных о потреблении кислорода и pH на микропланшетах. Методы Enzymol (2014) 547: 309–54.DOI: 10.1016 / B978-0-12-801415-8.00016-3
PubMed Реферат | CrossRef Полный текст | Google Scholar
12. Николас Д., Проктор Е.А., Раваль Ф.М., ИП Британская Колумбия, Хабиб С., Риту Е. и др. Достижения в количественной оценке митохондриальной функции в первичных иммунных клетках человека с помощью анализа внеклеточного потока. PloS One (2017) 12: e0170975. doi: 10.1371 / journal.pone.0170975
PubMed Аннотация | CrossRef Полный текст | Google Scholar
13. Дранка Б. П., Бенавидес Г. А., Диерс А. Р., Джордано С., Зеликсон Б. Р., Рейли С. и др.Оценка биоэнергетической функции в ответ на окислительный стресс с помощью метаболического профилирования. Free Radic Biol Med (2011) 51: 1621–35. doi: 10.1016 / j.freeradbiomed.2011.08.005
PubMed Аннотация | CrossRef Полный текст | Google Scholar
14. Чако Б.К., Крамер П.А., Рави С., Бенавидес Г.А., Митчелл Т., Дранка Б.П. и др. Индекс биоэнергетического здоровья: новая концепция в митохондриальных трансляционных исследованиях. Clin Sci (2014) 127: 367–73. doi: 10.1042 / CS20140101
CrossRef Полный текст | Google Scholar
15.Dimroth P, Kaim G, Matthey U. Решающая роль мембранного потенциала для синтеза АТФ с помощью F (1) F (o) АТФ-синтаз. J Exp Biol (2000) 203 (Pt 1): 51–9.
PubMed Аннотация | Google Scholar
21. Ан Б. Х., Ким Х. С., Сонг С., Ли И. Х., Лю Дж., Вассилопулос А. и др. Роль митохондриальной деацетилазы Sirt3 в регулировании энергетического гомеостаза. Proc Natl Acad Sci (2008) 105: 14447–52. DOI: 10.1073 / pnas.08037
PubMed Аннотация | CrossRef Полный текст | Google Scholar
22.Цимен Х, Хан МДж, Ян И, Тонг Кью, Коч Х, Коч Э. Регулирование активности сукцинатдегидрогеназы с помощью SIRT3 в митохондриях млекопитающих. Биохимия (2010) 49: 304–11. doi: 10.1021 / bi
PubMed Аннотация | CrossRef Полный текст | Google Scholar
25. Brooks GA, Dubouchaud H, Brown M, Sicurello JP, Butz CE. Роль митохондриальной лактатдегидрогеназы и окисления лактата во внутриклеточном лактатном челноке. Proc Natl Acad Sci USA (1999) 96: 1129–34.doi: 10.1073 / pnas.96.3.1129
PubMed Аннотация | CrossRef Полный текст | Google Scholar
26. Sugden MC, Holness MJ. Последние достижения в механизмах регуляции окисления глюкозы на уровне пируватдегидрогеназного комплекса с помощью PDK. Am J Physiol Endocrinol Metab (2003) 284: 855–62. doi: 10.1152 / ajpendo.00526.2002
CrossRef Полный текст | Google Scholar
27. Mimic-Oka J, Simic T, Djukanovic L, Reljic Z, Davicevic Z. Изменение антиоксидантной способности плазмы при различных степенях хронической почечной недостаточности. Clin Nephrol (1999) 51: 233–41.
PubMed Аннотация | Google Scholar
30. Hill BG, Benavides GA, Lancaster JR Jr, Ballinger S, Dell’Italia L, Jianhua Z, et al. Интеграция клеточной биоэнергетики с контролем качества митохондрий и аутофагией. Biol Chem (2012) 393: 1485–512. doi: 10.1515 / hsz-2012-0198
PubMed Аннотация | CrossRef Полный текст | Google Scholar
33. Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD.Вклад дыхания и гликолиза в выработку внеклеточной кислоты. Biochim Biophys Acta (2015) 1847: 171–81. doi: 10.1016 / j.bbabio.2014.10.005
PubMed Аннотация | CrossRef Полный текст | Google Scholar
34. Ву М., Нилсон А., Свифт А.Л., Моран Р., Тамагнин Дж., Парслоу Д. и др. Многопараметрический метаболический анализ показывает тесную связь между ослаблением биоэнергетической функции митохондрий и повышенной зависимостью от гликолиза в опухолевых клетках человека. Am J Physiol Cell Physiol (2007) 292: C125–36.doi: 10.1152 / ajpcell.00247.2006
PubMed Аннотация | CrossRef Полный текст | Google Scholar
35. Лант SY, Vander Heiden MG. Аэробный гликолиз: удовлетворение метаболических требований, связанных с пролиферацией клеток. Annu Rev Cell Dev Biol (2011) 27: 441–64. doi: 10.1146 / annurev-cellbio-092910-154237
PubMed Аннотация | CrossRef Полный текст | Google Scholar
40. Сен-Пьер Дж., Дрори С., Улдри М., Сильваджи Дж. М., Ри Дж., Джегер С. и др. Подавление активных форм кислорода и нейродегенерация коактиваторами транскрипции PGC-1. Cell (2006) 127: 397–408. doi: 10.1016 / j.cell.2006.09.024
PubMed Аннотация | CrossRef Полный текст | Google Scholar
41. Jäger S, Handschin C, St-Pierre J, Spiegelman BM. Действие AMP-активированной протеинкиназы (AMPK) в скелетных мышцах посредством прямого фосфорилирования PGC-1альфа. Proc Natl Acad Sci USA (2007) 104: 12017–22. doi: 10.1073 / pnas.0705070104
PubMed Аннотация | CrossRef Полный текст | Google Scholar
42. Заза Г., Граната С., Масола В., Ругиу К., Фантин Ф., Джезуальдо Л. и др.Подавление ядерно-кодируемых генов окислительного метаболизма у диализованных пациентов с хронической болезнью почек. PloS One (2013) 8: e77847. doi: 10.1371 / journal.pone.0077847
PubMed Аннотация | CrossRef Полный текст | Google Scholar
43. Карденес Н., Кори С., Гири Л., Джайн С., Жариков С., Баржа С. и др. Биоэнергетический скрининг тромбоцитов у пациентов с серповидными клетками выявляет ингибирование митохондриального комплекса V, что способствует активации тромбоцитов. Кровь (2014) 123: 2864–72.doi: 10.1182 / blood-2013-09-529420
PubMed Аннотация | CrossRef Полный текст | Google Scholar
44. Холливелл Б., Гаттеридж Дж. М.. Свободные кислородные радикалы и железо применительно к биологии и медицине: некоторые проблемы и концепции. Arch Biochem Biophys (1986) 246: 501–14. DOI: 10.1016 / 0003-9861 (86)
-X
PubMed Аннотация | CrossRef Полный текст | Google Scholar
46. Akiyama S, Inagaki M, Tsuji M, Gotoh H, Gotoh T, Gotoh Y, et al. Исследование мРНК индукции Cu / Zn-супероксиддисмутазы при гемодиализе. Nephron Clin Pract (2005) 99: c107–14. doi: 10.1159 / 000083928
PubMed Аннотация | CrossRef Полный текст | Google Scholar
47. Граната С., Заза Г., Симоне С., Виллани Г., Латорре Д., Понтрелли П. и др. Нарушение регуляции митохондрий и окислительный стресс у пациентов с хронической болезнью почек. BMC Genomics (2009) 10: 388. doi: 10.1186 / 1471-2164-10-388
PubMed Аннотация | CrossRef Полный текст | Google Scholar
49. Zhang R, Saredy J, Shao Y, Yao T., Liu L, Saaoud F, et al.Терминальная стадия почечной недостаточности отличается от хронической почечной недостаточности повышающей регуляцией провоспалительного секретома, модулируемого ROS, в PBMC — новой модели множественных попаданий для прогрессирования заболевания. Редокс Биол (2020) 34: 101460. doi: 10.1016 / j.redox.2020.101460
PubMed Аннотация | CrossRef Полный текст | Google Scholar
50. Рабинович Р.С., Самборска Б., Фауберт Б., Ма Э. Х., Гравий С.П., Анджеевский С. и др. AMPK поддерживает клеточный метаболический гомеостаз за счет регуляции митохондриальных активных форм кислорода. Cell Rep (2017) 21: 1–9. doi: 10.1016 / j.celrep.2017.09.026
PubMed Аннотация | CrossRef Полный текст | Google Scholar
52. Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. Деление митохондрий способствует дисфункции митохондрий и инсулинорезистентности скелетных мышц. Mol Cell Biol (2012) 32: 309–19. doi: 10.1128 / MCB.05603-11
PubMed Аннотация | CrossRef Полный текст | Google Scholar
53. Yu T, Sheu SS, Robotham JL, Yoon Y.Деление митохондрий опосредует гибель клеток, вызванную высоким уровнем глюкозы, за счет повышенного производства активных форм кислорода. Cardiovasc Res (2008) 79: 341–51. doi: 10.1093 / cvr / cvn104
PubMed Аннотация | CrossRef Полный текст | Google Scholar
54. Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q и др. Связанная с динамином GTPase Dnm1 регулирует деление митохондрий у дрожжей. Nat Cell Biol (1999) 1: 298–304. doi: 10.1038 / 13014
PubMed Аннотация | CrossRef Полный текст | Google Scholar
55.Лабрусс AM, Заппатерра MD, Рубе Д.А., ван дер Блик AM. Связанный с динамином белок DRP-1 C. elegans контролирует разрыв внешней мембраны митохондрий. Mol Cell (1999) 4: 815–26. doi: 10.1016 / S1097-2765 (00) 80391-3
PubMed Аннотация | CrossRef Полный текст | Google Scholar
56. Александр Ч., Вотруба М., Пеш У. Э., Тиселтон Д. Л., Майер С., Мур А. и др. OPA1, кодирующий связанную с динамином GTPase, мутирует при аутосомно-доминантной атрофии зрительного нерва, связанной с хромосомой 3q28. Нат Генет (2000) 26: 211–5.doi: 10.1038 / 79944
PubMed Аннотация | CrossRef Полный текст | Google Scholar
57. Делеттр С., Ленарс Дж., Гриффоин Дж. М., Гигарель Н., Лоренцо С., Беленгер П. и др. Ядерный ген OPA1, кодирующий митохондриальный белок, связанный с динамином, мутировал при доминантной атрофии зрительного нерва. Нат Генет (2000) 26: 207–10. doi: 10.1038 / 79936
PubMed Аннотация | CrossRef Полный текст | Google Scholar
58. Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, et al.АМФ-активированная протеинкиназа опосредует деление митохондрий в ответ на энергетический стресс. Science (2016) 351: 275–81. doi: 10.1126 / science.aab4138
PubMed Аннотация | CrossRef Полный текст | Google Scholar
59. Чако Б.К., Крамер П.А., Рави С., Джонсон М.С., Харди Р.В., Баллинджер С.В. и др. Методы определения различных биоэнергетических профилей тромбоцитов, лимфоцитов, моноцитов и нейтрофилов, а также окислительного выброса в крови человека. Лабораторное исследование (2013) 93: 690–700.doi: 10.1038 / labinvest.2013.53
PubMed Аннотация | CrossRef Полный текст | Google Scholar
61. Maynard S, Hejl AM, Dinh TST, Keijzers G, Hansen AM, Desler C, et al. Нарушение митохондриального дыхания, измененные пулы dNTP и снижение активности AP-эндонуклеазы 1 в мононуклеарных клетках периферической крови пациентов с болезнью Альцгеймера. Aging (Олбани, штат Нью-Йорк) (2015) 7: 793–810. doi: 10.18632 / age.100810
PubMed Аннотация | CrossRef Полный текст | Google Scholar
62.Jang DH, Greenwood JC, Spyres MB, Eckmann DM. Измерение митохондриального дыхания и подвижности при неотложной помощи: сепсис, травмы и отравления. J Intensive Care Med (2017) 32: 86–94. doi: 10.1177 / 0885066616658449
PubMed Аннотация | CrossRef Полный текст | Google Scholar
63. Кин К.Н., Калтон Е.К., Крузат В.Ф., Соарес М.Дж., Ньюсхолм П. Влияние криоконсервации на биоэнергетику лейкоцитов периферической крови человека. Clin Sci (Лондон) (2015) 128: 723–33. DOI: 10.1042 / CS20140725
PubMed Аннотация | CrossRef Полный текст | Google Scholar
64. Weinberg A, Louzao R, Mussi-Pinhata MM, Cruz MLS, Pinto JA, Huff MF, et al. Программа обеспечения качества криоконсервации мононуклеарных клеток периферической крови. Clin Vaccine Immunol (2007) 14: 1242–4. doi: 10.1128 / CVI.00187-07
PubMed Аннотация | CrossRef Полный текст | Google Scholar
65. Азиз Н., Марголик Дж. Б., Детелс Р., Ринальдо С. Р., Фаир Дж., Джеймисон Б. Д. и др. Значение программы оценки качества для оптимизации криоконсервации мононуклеарных клеток периферической крови в многоцентровом исследовании. Clin Vaccine Immunol (2013) 20: 590–5. doi: 10.1128 / CVI.00693-12
PubMed Аннотация | CrossRef Полный текст | Google Scholar
66. Холл А.М., Анвин Р.Дж. Не такой уж и «могучий хондрион»: возникновение заболеваний почек из-за митохондриальной дисфункции. Nephron Physiol (2007) 105: 1–10. doi: 10.1159 / 000096860
CrossRef Полный текст | Google Scholar
Трансэтническое менделевское рандомизированное исследование выявляет причинно-следственные связи между кардиометаболическими факторами и хроническим заболеванием почек
Резюме
ИСТОРИЯ ВОПРОСА Бремя хронической болезни почек (ХБП) для общественного здравоохранения является значительным и не снизилось, как ожидалось, с текущими вмешательства по лечению болезней.С ХБП связано большое количество клинических, биологических и поведенческих факторов риска. Однако неясно, какие из них являются причинными.
ЦЕЛЬ Систематически проверять, связаны ли ранее сообщенные факторы риска ХБП с этим заболеванием у европейских и восточноазиатских предков.
DESIGN Менделирующая рандомизация (MR) по двум выборкам и нелинейный MR-анализ.
УЧАСТНИКИ 53 703 случая CKD и 960 624 контрольных случая европейского происхождения от CKDGen, UK Biobank и HUNT, а также 13 480 случаев CKD и 238 118 контрольных образцов восточноазиатского происхождения от Biobank Japan, China Kadoorie Biobank и Japan-Kidney-Biobank / ToMMo.
МЕРЫ Систематический анализ литературы исследований PubMed выявил 45 клинических факторов риска и биомаркеров с надежно связанными генетическими вариантами, включая фенотипы, связанные с артериальным давлением, диабетом, глюкозой, инсулином, липидами, ожирением, курением, нарушениями сна, нефролитиазом, мочевой кислотой. , ишемическая болезнь сердца, минеральная плотность костей, гомоцистеин, С-реактивный белок, питательные микроэлементы и функция щитовидной железы, которые были выбраны в качестве воздействия. Результатом была ХБП (определяемая клиническим диагнозом или расчетной скоростью клубочковой фильтрации (рСКФ) <60 мл / мин / л.73м 2 ).
РЕЗУЛЬТАТЫ Восемь факторов риска продемонстрировали доказательства причинного воздействия на ХБП у европейцев, включая индекс массы тела (ИМТ), гипертонию, систолическое артериальное давление, холестерин липопротеинов высокой плотности, аполипопротеин AI, липопротеин A, диабет 2 типа (T2D). и нефролитиаз. У восточноазиатских предков ИМТ, СД2 и нефролитиаз показали наличие причинных эффектов на ХБП. Гипертония продемонстрировала надежные доказательства сильного причинного эффекта на ХБП у европейцев, но, напротив, показала нулевой эффект у жителей Восточной Азии, что предполагает возможность различных причинных факторов риска у европейцев и жителей Восточной Азии.Хотя предрасположенность к СД2 продемонстрировала стойкое влияние на ХБП, влияние гликемических характеристик на ХБП было слабым, что позволяет предположить, что СД2 может иметь независимые от глюкозы механизмы влияния на ХБП. Нелинейная МРТ указала на пороговую взаимосвязь между генетически предсказанным ИМТ и ХБП с повышенным риском при ИМТ выше 25 кг / м 2 .
ОГРАНИЧЕНИЕ Из-за несбалансированного распределения данных между родословными, мы смогли проверить только 17 из 45 факторов риска у участников из Восточной Азии.
ВЫВОДЫ Восемь факторов риска, связанных с ХБП, продемонстрировали причинное влияние на заболевание у более чем 1,2 миллиона предков в Европе и Восточной Азии. Эти факторы риска были в основном связаны с кардио-метаболическим здоровьем, что подтверждает общую причинную связь между кардио-метаболическим здоровьем и функцией почек. Это исследование предоставляет доказательства потенциальных целей вмешательства для первичной профилактики ХБП, которые могут помочь снизить глобальное бремя ХБП и сопутствующих сердечно-метаболических заболеваний.
Доказательства до этого исследования Хроническая болезнь почек (ХБП) оказывает серьезное влияние на глобальное здоровье, как прямая причина заболеваемости и смертности, так и как важное осложнение сердечно-метаболических заболеваний. Однако даже при существующих вмешательствах бремя ХБП не снизилось, как ожидалось, за последние 30 лет. Существующие эпидемиологические исследования ХБП в основном сосредоточены на лечении заболеваний у пациентов из определенных групп населения и оценке связи, а не причинно-следственной связи.Срочно необходима систематическая оценка причинных детерминант ХБП в различных популяциях, чтобы способствовать переходу от лечения пациентов с ХБП к профилактике заболевания в группах высокого риска. Использование генетических данных и новейших методологий менделевской рандомизации (MR) предлагает рентабельный способ оценки потенциальных целей вмешательства для профилактики ХБП в группах высокого риска.
Дополнительная ценность этого исследования В этом исследовании мы систематически построили причинный атлас 45 факторов риска ХБП у европейских и восточноазиатских предков с использованием МРТ.Чтобы максимизировать эффективность этих анализов и точность результатов, мы собрали и согласовали данные генетической ассоциации ХБП из шести крупных биобанков (более 1,1 миллиона европейцев и 250 000 жителей Восточной Азии). Применяя комплексную структуру МРТ, включая линейную МРТ с двумя выборками, двунаправленную МРТ, многомерную МРТ и нелинейную МРТ, мы идентифицировали восемь факторов риска с надежными доказательствами причинного воздействия на ХБП в европейских исследованиях происхождения, включая индекс массы тела (ИМТ). ), гипертония, систолическое артериальное давление, холестерин липопротеинов высокой плотности, аполипопротеин AI, липопротеин A, диабет 2 типа (T2D) и нефролитиаз.В восточноазиатских исследованиях ИМТ, СД2 и нефролитиаз также показали причинное влияние на ХБП. Среди других факторов, гипертония показала надежные доказательства сильного причинного эффекта на ХБП у европейцев, но, напротив, у жителей Восточной Азии не показала никакого эффекта. Это открытие МРТ вместе с предыдущими литературными данными открывает возможность того, что гипертония может играть разные причинные роли в ХБП у разных предков. Для диабета и гликемических фенотипов наши анализы MR и чувствительности предполагают причинную роль предрасположенности T2D к CKD, но указывают на слабое влияние гликемических фенотипов на CKD.Это согласуется с недавним исследованием ингибиторов SGLT2 при заболевании почек, из которого следует, что СД2 может иметь независимые от глюкозы механизмы влияния на ХЗП. Что касается липидных фенотипов, мы нашли убедительные доказательства, подтверждающие роль холестерина липопротеинов высокой плотности в ХЗП, а также предположили влияние двух липидных мишеней: уровня циркулирующего СЕТР и концентрации липопротеина А. Для массы тела в нашем исследовании была определена пороговая взаимосвязь между ИМТ и ХБП с повышенным риском при ИМТ выше 25 кг / м 2 .О причинно-следственной связи между нефролитиазом и ХБП сообщалось в предыдущих исследованиях, но наше исследование впервые подтвердило причинную связь между ними.
Применение всех доступных доказательств Это исследование является значительным шагом вперед в установлении приоритетных целевых показателей вмешательства при ХБП для более чем 1,2 миллиона участников. В нашем исследовании представлены причинно-следственные доказательства из выборок как европейского, так и восточноазиатского населения, что расширяет возможность обобщения причинно-следственного атласа.Важно отметить, что приоритетные факторы риска в основном связаны с сердечно-метаболическим здоровьем, что поддерживает общую причинно-следственную связь между сердечно-метаболическим здоровьем и функцией почек. С клинической точки зрения высококачественные доказательства этого исследования подчеркивают важность изучения этих причинных факторов в общей популяции и уделяют приоритетное внимание целевым показателям лекарственных препаратов и вмешательствам, связанным с образом жизни, для первичной профилактики ХБП, что может помочь снизить глобальное бремя ХБП и ее сердечно-метаболические заболевания. сопутствующие заболевания.
Заявление о конкурирующем интересе
Авторы заявили об отсутствии конкурирующего интереса.
Отчет о финансировании
JZ финансируется стипендией вице-канцлера Бристольского университета. Это исследование также финансировалось отделом интегративной эпидемиологии Совета медицинских исследований Великобритании (MC_UU_00011 / 1, MC_UU_00011 / 4 и MC_UU_00011 / 7). Это исследование финансировалось / поддерживалось Центром биомедицинских исследований NIHR при университетских больницах Bristol NHS Foundation Trust и Бристольским университетом (GDS, TRG и REW). Это исследование получило финансирование от Совета медицинских исследований Великобритании (MR / R013942 / 1).Мнения, выраженные в этой публикации, принадлежат автору (авторам) и не обязательно принадлежат NHS, Национальному институту исследований в области здравоохранения или Департаменту здравоохранения и социальной защиты. YMZ поддерживается Национальным фондом естественных наук Китая (81800636) и Пекинским фондом естественных наук (7184253). HZ поддерживается программой обучения основного плана исследований Национального фонда естественных наук Китая (120), грантом Пекинского научно-технологического проекта (D18110700010000) и системой здравоохранения Мичиганского университета. Медицинские науки Пекинского университета. Центр Объединенного института трансляционных и клинических исследований (BMU2017JI007).NF поддерживается наградами Национального института здравоохранения R01-MD012765, R01-DK117445 и R21-HL140385. RC финансируется стипендиатом по клинической академической подготовке Wellcome Trust GW4 (WT 212557 / Z / 18 / Z). Исследование здоровья Норд Тронделаг (Исследование HUNT) является результатом сотрудничества исследовательского центра HUNT (факультет медицины и медицинских наук, NTNU, Норвежский университет науки и технологий), Совета округа Норд Тронделаг, Регионального управления здравоохранения Центральной Норвегии и Норвежского института. общественного здравоохранения.MCB поддерживается стипендиатом Совета по медицинским исследованиям Великобритании (MRC) по развитию навыков (MR / P014054 / 1). SF поддерживается докторской степенью Wellcome Trust (WT108902 / Z / 15 / Z). QY финансируется стипендией PhD Китайского совета по стипендиям (CSC201808060273). YC был поддержан Национальной ключевой программой исследований и разработок Китая (2016YFC0
0, 2016YFC0
1, 2016YFC0
4). Базовое исследование China Kadoorie Biobank и первое повторное исследование были поддержаны грантом благотворительного фонда Kadoorie Charitable Foundation в Гонконге.Долгосрочное наблюдение поддерживается грантами UK Wellcome Trust (202922 / Z / 16 / Z, 088158 / Z / 09 / Z, 104085 / Z / 14 / Z). Japan-Kidney-Biobank получил поддержку AMED в рамках гранта № 20km0405210. PCH поддерживается Cancer Research UK [номер гранта: C18281 / A19169]. АК поддерживался ДФГ КО 3598 / 5-1. Н.Ф. поддерживается наградами NIH R01-DK117445, R01-MD012765, R21-HL140385.
Заявления авторов
Я подтверждаю, что были соблюдены все соответствующие этические принципы и получены все необходимые разрешения IRB и / или комитета по этике.
Да
Подробная информация об IRB / надзорном органе, предоставившем одобрение или исключение для описанного исследования, приведена ниже:
В документе использовалась только сводная статистика из общественного достояния, поэтому одобрения по этническим вопросам относятся к исходным исследованиям GWAS
Все Получено необходимое согласие пациента / участника и заархивированы соответствующие институциональные формы.
Да
Я понимаю, что все клинические испытания и любые другие проспективные интервенционные исследования должны быть зарегистрированы в одобренном ICMJE реестре, таком как ClinicalTrials.губ. Я подтверждаю, что любое такое исследование, указанное в рукописи, было зарегистрировано и предоставлен идентификатор регистрации исследования (примечание: при публикации проспективного исследования, зарегистрированного ретроспективно, просьба предоставить заявление в поле идентификатора исследования, объясняющее, почему исследование не было зарегистрировано заранее) .
Да
Я выполнил все соответствующие инструкции по составлению отчетов об исследованиях и загрузил соответствующие контрольные списки отчетов по исследованиям сети EQUATOR и другие соответствующие материалы в качестве дополнительных файлов, если применимо.
Да
Фредрик Палм — Университет Упсалы, Швеция
Физиология почек и болезни почек
Болезнь почек — основная причина преждевременной смертности. В последнее время акцент сместился с гломерулоцентрического на преимущественно тубулоцентрический взгляд на механизмы, участвующие в развитии заболевания почек, вызванного диабетом, гипертонией или острым повреждением почек (Vallon and Thomson 2012). Наша исследовательская программа направлена на лучшее понимание базовой физиологии почек и патологических механизмов, ведущих к заболеванию почек.В последнее время мы приложили значительные усилия для определения участия нарушенного кислородного гомеостаза почек в возникновении и прогрессировании острого и хронического заболевания почек.
Напряжение кислорода в ткани почек низкое уже в нормальных условиях (Aukland and Krog 1960, Leichtweiss et al 1969), и попытки увеличить доставку кислорода за счет увеличения почечного кровотока обычно также приводят к увеличению канальцевой нагрузки электролитов из-за повышенной скорости клубочковой фильтрации, которая сам по себе увеличивает метаболические потребности.Таким образом, любое усиление метаболизма в почках может привести к снижению напряжения кислорода в ткани почек, то есть к гипоксии. Действительно, повышенный метаболизм почек связан с диабетической нефропатией (Korner et al, 1994), а диабет связан со снижением давления кислорода в ткани почек как у животных, так и у пациентов (Ries et al 2003, dos Santos et al 2007, Rosenberger et al 2008, Edlund и др., 2009 г., Хайдара и др., 2009 г., Иноуэ и др., 2011 г.). Fine et al. предположили, что начальное повреждение клубочков снижает кровоток через перитубулярные капилляры и приводит к снижению оксигенации почек, способствуя тубулоинтерстициальному фиброзу и прогрессированию поражения почек (Fine, Orphanides et al., 1998).Важно отметить, что хроническая тубулоинтерстициальная гипоксия была предложена в качестве общего пути к конечной стадии почечной недостаточности независимо от начального инсульта (Nangaku 2004, Nangaku 2006, Singh et al 2008, Mimura and Nangaku 2010, Palm and Nordquist 2011).
В 2003 году мы представили наш первый отчет, демонстрирующий внутрипочечную тканевую гипоксию при диабете (Palm et al. 2003), который после этого был подтвержден несколькими международными лабораториями (Ries et al 2003, dos Santos et al 2007, Rosenberger et al 2008, Инь и др., 2012).С тех пор мы продолжаем исследовать роль гипоксии почечной ткани в развитии диабетической нефропатии, а также при других состояниях, связанных с повышенным риском заболевания почек, таких как гипертензия, ишемия-реперфузия и хирургическая нефрэктомия 5/6.
Рисунок. Объединяющая гипотеза деятельности лаборатории. Большинство путей уже опубликовано. Некоторые из путей, перечисленных в этой общей схеме, в настоящее время поддерживаются только неопубликованными данными, но вскоре будут опубликованы.
Участие связанных с воспалением miR-155 и miR-146a в диабетической нефропатии: последствия для повреждения эндотелия клубочков | BMC Nephrology
Подготовка образцов почек человека
В это исследование были включены шесть пациентов с DN 2 типа (две женщины и четыре мужчины, возраст 51,7 ± 7,8 года), ткани почек были взяты из биопсии почек. Клиническая стадия почек и патологическая классификация этих шести пациентов были отнесены к стадии DN по Mogosen [20] и патологической классификации DN, установленной Обществом патологов почек [21].Трое пациентов, у которых была диагностирована карцинома почек и которые подвергались нефрэктомии, но у которых не было выявлено гипертонии, диабета и других сопутствующих заболеваний, были включены в контрольную группу (одна женщина и двое мужчин, возраст 44 ± 12,9 года, анализ мочи и креатинин сыворотки были нормальными) (Таблица 1). OGTT трех контрольных групп, включенных в наше исследование, был нормальным, поэтому мы думали, что это не диабет. Ткани почек получали в результате биопсии почек (у пациентов) или после операции по нефрэктомии (в контрольной группе, в основном коры почек) после получения подписанного информированного согласия от людей.Ткани использовали для микроматрицы miRNA и полимеразной цепной реакции (ПЦР) в реальном времени или фиксировали в формалине и заключали в парафин для гистологической гибридизации и гибридизации in situ. Изменения морфологии почек исследовали в формалине, парафиновых срезах ткани (4 мкм), окрашенных Periodic Acid-Schiff (PAS). Исследование было одобрено Комитетом по этике исследований на людях Сычуаньского университета (Сычуань, Китай).
Таблица 1
Демография пациентов с DN
Микроматрица
MicroRNA и анализ данных
Рассеченная ткань почек была гомогенизирована и РНК экстрагирована с использованием TRIzol (Invitrogen, Carlsbad, CA, USA) в соответствии с протоколом производителя.РНК очищали с использованием мини-набора miRNeasy (QIAGEN) в соответствии с инструкциями производителя. Качество и количество РНК определяли с помощью Nanodrop 1000. Чтобы изучить экспрессию miRNA у пациентов с диабетом, мы выполнили профилирование экспрессии miRNA в образцах почек от шести пациентов с диабетом и трех здоровых контролей с использованием микрочипов miRNA (Exiqon, Vedbaek, Дания). ) согласно инструкции производителя; сервис микрочипов miRNA был предоставлен KangChen Bio-Tech, Inc., Шанхай, Китай. Вкратце, после выделения РНК использовали набор для маркировки miRCURY ™ Hy3 ™ / Hy5 ™ Power (Exiqon, Vedbaek, Дания) в соответствии с инструкциями производителя по маркировке miRNA. Один микрограмм каждого образца был помечен на 3’-конце флуоресцентной меткой Hy3TM путем объединения РНК (один микрограмм) в 2,0 мкл воды с 1,0 мкл буфера CIP (0,5 мкл) и CIP (0,5 мкл) (Exiqon). Смесь инкубировали 30 мин при 37 ° C, и реакцию останавливали инкубацией в течение 5 мин при 95 ° C.Затем 3,0 мкл буфера для маркировки, 1,5 мкл флуоресцентной метки (Hy3TM), 2,0 мкл DMSO и 2,0 мкл фермента для маркировки (этот фермент на самом деле был ферментом для маркировки из набора для маркировки miRCURYTM Array Power (Cat # 208032-A, Exiqon)). Реакцию мечения инкубировали в течение 1 ч при 16 ° C, и реакцию останавливали инкубацией в течение 15 мин при 65 ° C. Меченый Hy3TM образец (25 мкл) с 25 мкл гибридизационного буфера денатурировали в течение 2 минут при 95 ° C, инкубировали на льду в течение 2 минут, а затем гибридизовали с miRCURYTM LNA Array (v.16.0) (Exiqon) в соответствии с инструкциями производителя в течение 16-20 часов при 56 ° C в системе гибридизации с 12 отсеками (Hybridization System; Nimblegen Systems, Inc., Мэдисон, Висконсин, США). После гибридизации слайды несколько раз промывали с использованием набора промывочного буфера (Exiqon) и сушили центрифугированием в течение 5 минут при 400 об / мин (слайды сушили центрифугированием в течение 5 минут при 1000 об / мин). Затем слайды сканировали с помощью сканера микрочипов Axon GenePix 4000B (Axon Instruments, Фостер-Сити, Калифорния, США), и изображения импортировали с помощью GenePix Pro 6.0 (Axon Instruments) для выравнивания сетки и извлечения данных. Реплицированные miRNA усредняли, и для расчета коэффициента нормализации отбирали те, которые имели интенсивность ≥50. После нормализации значительные дифференциально экспрессируемые miRNA были идентифицированы с помощью фильтрации Volcano Plot. Иерархическая кластеризация выполнялась с использованием программного обеспечения MEV (v4.6, TIGR) (рисунок 1).
Рисунок 1
Иерархическая кластеризация для дифференциально экспрессируемых miRNA в DN по сравнению с контролем. Тепловая карта была сгенерирована неконтролируемым иерархическим кластеризацией профилей miRNA 6 пациентов с диабетом и 3 здоровых людей из контрольной группы. Порог, который мы использовали для скрининга микроРНК с повышением или понижением регуляции, был кратным изменением ≥ 2,0. Легенда тепловой карты: красный = регулирование с повышением, зеленый = регулирование с понижением. Строки генов и столбцов представляют образец.
Гибридизация in situ
Осуществлялось обнаружение in situ миРНК in situ на основе заблокированных нуклеиновых кислот в срезах почек. Вкратце, образцы почек фиксировали при 4 ° C в формалине, заливали парафином и разрезали на срезы 4 мкм.Зонды, меченные Dig, были сконструированы и синтезированы Invitrogen Inc. Последовательности меченных Dig зондов против мРНК miR-155 и miR-146a были следующими: miR-155: 5′-CCCCTATCACGATTAGCATTAA-3 ‘, miR-146a: 5 ′ -AACCCATGGAATTCAGTTCTCA-3 ′. Скремблированную последовательность зонда: 5’-TTCACAATGCGTTATCGGATGT-3 ‘использовали в качестве отрицательного контроля для сигнала неспецифической гибридизации. Гибридизацию in situ проводили с использованием набора для обнаружения на срезах тканей почек, фиксированных 4% параформальдегидом (содержащих 0,1% диэтилпирокарбоната), в соответствии с инструкциями производителя.
Модели крыс
Однопометные крысы Sprague Dawley (SD) (самцы, возраст 6 недель, 200–220 г) содержались в стандартных лабораторных условиях при температуре 22 ± 2 ° C с 12-часовым освещением / 12-часовым освещением. h темный цикл и относительная влажность 40% –60%. Их разделили на две группы: крысы без диабета (контроль, n = 6) и крысы с индуцированным стрептозотоцином (STZ) диабетом 1 типа (DM, n = 18). Модели крыс с СД индуцировали внутрибрюшинной инъекцией однократной дозы 60 мг / кг СТЗ (Sigma, St.Луис, Миссури, США) в цитратном буфере (pH 4,5). Случайный уровень глюкозы в крови тестировали с помощью глюкометра (Accu-Chek, Roche, Базель, Швейцария), и уровни глюкозы в крови определяли на 3 и 7 дни после инъекции STZ. В исследовании использовались только крысы с концентрацией глюкозы ≥16,7 ммоль / л в течение 3 дней подряд. Крыс DM подвергали эвтаназии через 1, 4 и 8 недели после индукции диабета и извлекали их почки. Их кору почек собирали для ПЦР в реальном времени путем тщательного удаления почечной лоханки и мозгового вещества.Протокол эксперимента был одобрен Комитетом по этике экспериментов на животных Сычуаньского университета.
В отдельном эксперименте по созданию крыс SD диабета 2 типа крысы (самцы, в возрасте 6 недель, 200–220 г) были назначены на один из двух режимов питания в течение начального периода в 6 недель: нормальный гранулированный рацион (NPD, n = 6) или диета с высоким содержанием жиров и сахара (HFD, содержащая обычную диету плюс 27,3% сала, 54,6% сахарозы, 16,4% холестерина и 1,6% холата натрия (масс.), n = 18).По истечении этого времени животные HFD с высоким HOMA-IR (глюкоза в плазме натощак (ммоль / л) × инсулин натощак (мМЕ / л) ÷ 22,5) были определены как инсулинорезистентные, и им вводили несколько низких доз STZ (четыре дозы 25 мг / кг; Sigma, Сент-Луис, Миссури, США) в цитратном буфере (pH 4,5) после ночного голодания. Животным NPD вводили цитратный буфер (1 мл / кг). Для исследования использовали крыс с уровнем глюкозы натощак ≥16,7 ммоль / л через 72 часа после инъекции СТЗ в течение 3 дней подряд. Крыс содержали на соответствующей диете до конца исследования.Крыс DM умерщвляли через 0, 8 и 16 недель после индукции диабета, собирали их почки и собирали их кору почек, как и раньше. Все протоколы экспериментов с участием животных были одобрены Комитетом по этике экспериментов на животных Сычуаньского университета.
Биохимические измерения при диабете у крыс
Уровни глюкозы в крови натощак (6 часов) определяли еженедельно в течение первых 2 недель, а затем каждые 2 недели с помощью глюкометра (Optium ™ Xceed Systems, Виктория, Австралия).Перед индукцией заболевания (неделя 0) мочу собирали в течение 24 часов, помещая крыс в метаболические клетки и выполняя анализ экскреции альбумина с мочой. Этот процесс повторяли после индукции заболевания (для диабета 1 типа: недели 1, 4 и 8; для диабета типа 2: недели 0, 8 и 16). Альбумин мочи определяли с помощью конкурентного ELISA в соответствии с инструкциями производителя (MaxiSorp Surface Immuno Plates ™; Nunc, Роскилле, Дания). Креатинин сыворотки определяли с помощью набора QuantiChrom Creatinine Assay Kit (BioAssay Systems, Хейворд, Калифорния, США).
Количественная ПЦР в реальном времени для экспрессии miR-155 и miR146a в почках человека и крысы
Чтобы подтвердить результаты профилирования miRNA у людей, мы использовали количественную ПЦР в реальном времени для измерения экспрессии miR-155 и miR-146a как в почках человека, так и в почках крысы. В реакции обратной транскрипции использовали обратную транскриптазу MMLV (Epicenter, Madison, WI, USA), а количественную ПЦР проводили с использованием системы ABI PRISM7500 (Applied Biosystems, Foster City, CA, USA).miR-155 и miR-146a были дополнительно количественно определены с помощью количественной ПЦР в реальном времени TaqMan. Используемые праймеры для ПЦР показаны в таблице 2.
Таблица 2.
Последовательность праймера для ПЦР в реальном времени
Клеточная культура
Нормальные HRGEC были приобретены в ScienCell Research Laboratories (Карлсбад, Калифорния, США) и культивированы в среде эндотелиальных клеток (ScienCell), содержащей 5% фетальной бычьей сыворотки и 1% добавки для роста эндотелиальных клеток при 37 ° C в течение 5 дней. % CO 2 атмосферы.Клетки были последовательно пассированы кратковременным воздействием 0,25% трипсина (BD-Difco, США) и 0,04% EDTA (Sigma, Сент-Луис, Миссури, США), и эксперименты проводились на клетках на пассажах 2–5 при 80% слияние. Их высевали с плотностью 1 × 10 5 клеток / лунку в 6-луночные чашки или 2,5 × 10 5 клеток / 25 см в колбу 2 . Перед экспериментами клетки голодали в течение 24 часов в поддерживающей среде.
Чтобы обнаружить зависящие от времени изменения экспрессии miR-155 и miR-146a, клетки, культивированные со средами, содержащими 25 ммоль / л глюкозы, культивировали при 37 ° C в 5% CO 2 для 0,0.5, 1, 2, 4, 8 и 24 часа. Затем их собирали для ПЦР в реальном времени. Химически модифицированные олигонуклеотиды РНК, содержащие последовательность, комплементарную зрелым miR-155 и miR-146a (ингибиторы miR-155 и miR-146a), использовали для ингибирования активности miR-155 и miR-146a. Миметики miR-155 и miR-146a представляли собой двухцепочечные конструкции, состоящие из направляющей и пассажирской цепей. Олигонуклеотид, содержащий ошибочное спаривание из четырех оснований (ненаправленная скремблирующая РНК), использовали в качестве отрицательного контроля. Пирролидин дитиокарбамат (PDTC, Sigma) использовался в качестве ингибитора NF-κB.В соответствии с установленными временными точками, HRGEC (1 × 10 5 на лунку) были трансфицированы миметиками, ингибиторами miR-155 и miR-146a и скремблирующим контролем через 24 часа голодания в бессывороточной среде с использованием липофектамина 2000. (Invitrogen) в соответствии с инструкциями производителя.
Вестерн-блоттинг
Белки экстрагировали из HRGEC с использованием буфера для лизиса RIPA и анализировали вестерн-блоттингом, как описано ранее [17]. Вкратце, после блокирования неспецифического связывания 5% бычьим сывороточным альбумином мембраны инкубировали с первичными антителами против TNF-α и трансформирующего фактора роста (TGF) -β1 в течение ночи при 4 ° C.После промывки мембраны инкубировали с вторичными антителами, конъюгированными с IRDye 800 (Rockland Immunochemicals, Inc., Гилбертсвилл, Пенсильвания, США), и сигналы детектировали с помощью системы инфракрасной визуализации Odyssey (Li-COR Biosciences, Линкольн, Северная Каролина, США ) и количественно оценивали с помощью программного обеспечения Image J (NIH). Соотношение белков нормализовали по GAPDH и выражали как среднее значение ± стандартная ошибка среднего (SEM).
Количественная ПЦР в реальном времени для экспрессии мРНК в HRGEC
Суммарную РНК экстрагировали из HRGEC с использованием набора RNeasy в соответствии с инструкциями производителя (Qiagen Inc.