Диатез. Нервно-артритический диатез у детей: причины, симптомы, лечение
Каждый из нас уже рождается со склонностью к определенным болезням, что обусловлено совокупностью наследственных факторов, заболеваниями родителей и родственников. Именно эта предрасположенность и носит название диатез или аномалия конституции.
Одно из них — нервно-артритический диатез. О нем мы и поговорим…
Что такое нервно-артритический диатез?
В его основе лежит нарушение обмена некоторых веществ в организме. Для этого состояния характерны расстройства питания, склонность к кетоацидозу (увеличению кетоновых тел в крови), повышенная нервная возбудимость.
Частота нервно-артритического диатеза составляет около 5%, то есть он развивается значительно реже, чем все остальные аномалии конституции.
Наиболее часто он встречается у детей в возрасте от двух до десяти лет, а к подростковому возрасту все его симптомы, как правило, нивелируются.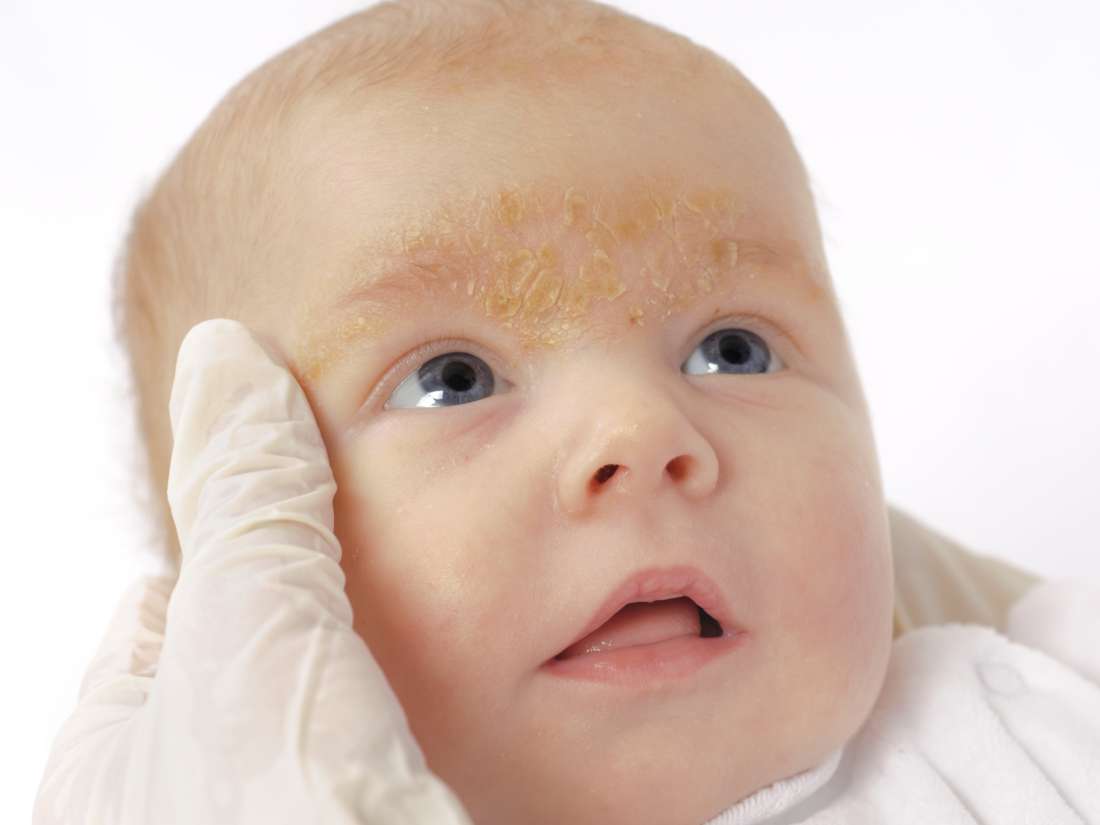 Однако в некоторых случаях у взрослых его проявления сохраняются на всю жизнь и со временем могут привести к развитию многих недугов: сахарному диабету, артериосклерозу, подагре, мочекаменной болезни и другим.
Однако в некоторых случаях у взрослых его проявления сохраняются на всю жизнь и со временем могут привести к развитию многих недугов: сахарному диабету, артериосклерозу, подагре, мочекаменной болезни и другим.
Причины
В основе диатеза лежит:
С одной стороны — наследственно обусловленный дефект обмена пуриновых оснований (веществ, содержащихся в мясе и некоторых других продуктах), а также усиленное образование мочевой кислоты в организме.
С другой стороны — воздействие факторов внешней среды: перегрузка рациона беременной женщины и ребенка раннего возраста продуктами, содержащими белки (мясо, печень, почки).
В таких семьях чаще выявляется неврастения, подагра, желчно или мочекаменная болезнь, артериосклероз, артериальная гипертензия, сахарный диабет.
Механизм развития
Довольно сложен. Однако, отбросив большинство сложных специфических терминов и не вдаваясь глубоко в патофизиологию, он сводится к следующему:
Однако, отбросив большинство сложных специфических терминов и не вдаваясь глубоко в патофизиологию, он сводится к следующему:
На фоне нарушенного обмена веществ под воздействием неблагоприятных факторов (стресс, погрешности в диете) в крови и моче повышается концентрация пуринов, мочевой кислоты, а также кетоновых тел (ацетон, ацетоуксусная и бетаоксимасляная кислота).
Все изменения, которые происходят в органах и системах крохи, обусловлены именно влиянием этих веществ.
Проявления
Зависят от возраста маленького пациента, а также выраженности симптомов диатеза. Выделяют несколько синдромов:
* Неврастенический синдром. Встречается наиболее часто и уже в грудном возрасте наблюдаются его симптомы: малыш беспричинно беспокоен и пуглив, плохо и мало спит. А по мере взросления кроха становится еще более возбудимым: может кричать даже от укуса комара или небольшого зуда кожи.
Поскольку пуриновые основания стимулируют нервную систему, кроха опережает своих сверстников в психическом и эмоциональном развитии. Малыш очень любознателен, быстро овладевает устной и письменной речью, легко запоминает прочитанное и услышанное. Такого ребенка зачастую называют вундеркиндом.
Однако это всего лишь внешнее благополучие. На самом деле проблема лежит гораздо глубже.
Нередко у ребенка старшего возраста с неврастеническим синдромом наблюдается:
- эмоциональная лабильность: неустойчивое настроение, повышенная раздражительность и плаксивость
- чрезмерная двигательная активность
- непродолжительный и неглубокий сон, ночные страхи
- повышенная чувствительность к запахам
- иногда привычные рвоты
- навязчивый кашель, тики
- энурез (ночное недержание мочи)
- стойкая анорексия, плохо поддающаяся лечению
- заикание (логоневроз)
- головные боли, а также боли в животе и мышцах
- обильный стул, несмотря на плохой аппетит
- по утрам запах ацетона (перезрелых фруктов) изо рта
Кроме того, ребенок с нервно-артритическим диатезом имеет небольшую массу тела. Однако иногда он склонен к полноте (особенно девочки).
Однако иногда он склонен к полноте (особенно девочки).
* Дисметаболический синдром. При этой форме, за счет нарушения обменных процессов, в организме ребенка происходят следующие изменения:
- Ураты во внутрисуставной жидкости могут кристаллизоваться. Поэтому иногда ребенок жалуется на ночные переходящие боли в суставах.
- Нередко у малыша в моче появляются соли (ураты, оксалаты, фосфаты) и эритроциты (за счет повреждения слизистой кристаллами соли). Поэтому ребенок может предъявлять жалобы на боли и жжение во время мочеиспускания, которые не связанны с переохлаждением или инфекцией.
Кроме того, при этом синдроме за счет резкого повышения в крови и тканях кетоновых тел иногда развивается ацетонемический криз. Его причинами могут стать погрешности в диете (злоупотребление мясными и жирными продуктами), любое заболевание или стресс, а также повышенная физическая нагрузка.
Симптомы ацетонемического криза:
* Внезапно или после кратковременного недомогания (головные боли, тошнота, запор, слегка серовато-белый цвет стула) появляется повторная неукротимая рвота и выраженный запах ацетона изо рта.
* Малыш жалуется на боли в животе, у него повышается температура тела, он может быстро терять в весе.
* Малютка отказывается от еды и питья.
* У крохи стремительно развиваются симптомы обезвоживания: сильная жажда и сухость во рту, сниженная эластичность кожи (если ее сильно сдавить, она медленно возвращается в нормальное положение), отсутствие слез.
* В тяжелых случаях иногда нарушается сознание и развиваются судороги.
Приступ продолжается от нескольких часов до одного-двух дней (иногда до пяти) и в большинстве случаев прекращается так же внезапно, как и начался. Обычно после приступа ребенок быстро идет на поправку.
Обычно после приступа ребенок быстро идет на поправку.
Изменения в анализах во время приступа
- В крови повышаются кетоновые тела, ацетон, аммиак и лейкоциты, а глюкоза, наоборот, может понижаться.
- В моче выявляется ацетон.
В случае развития выраженного ацетонемического криза ребенок нуждается в срочной медицинской помощи.
Спастический синдром
Для него характерно появление мигренеподобных головных болей, бронхоспазма, болей в области сердца, а также склонности к повышению артериального давления, запорам, почечным и кишечным коликам.
Возможно развитие бронхиальной астмы. Как правило, она протекает нетяжело и хорошо поддается лечению.
Кожный синдром
Развивается нечасто и в основном у детей старшего возраста. Он характеризуется развитием крапивницы, отёка Квинке, экземы, нейродермита.
Примите к сведению! Не всегда имеется четкая граница между этими синдромами. Поэтому, к примеру, у одного и того же ребенка могут наблюдаться признаки двух синдромов, но с преобладанием симптомов одного из них.
Когда необходимо лечение?
Ребенок нуждается в лечении в тяжелых случаях.
Для улучшения общего самочувствия назначаются:
- Успокоительные травы (валериана, мята, хвощ, пустырник) или препараты (фенобарбитал и другие).
- Лекарственные вещества, которые нарушают синтез мочевой кислоты (алопуринол) или выводят её из организма (этамид, уродан), препятствуют образованию кетоновых тел (пантотенат кальция).
Однако наиболее часто малыш нуждается в лечении при ацетонемическом кризе. Оно направленно на борьбу с обезвоживанием и ацидозом (состояние, при котором реакция крови кислая), ускорение выведения и разрушения кетоновых тел, а также прекращения рвоты.
Для этого рекомендуется:
- При невыраженном кризе (ацетон в моче не более «++») обильно поить ребенка. Подойдет сладкий чай, щелочные минеральные воды, свежеприготовленные фруктовые соки. Рекомендуется поить часто (каждые 10-15 минут) и небольшими порциями, чтобы не спровоцировать приступы повторной рвоты.
- Если симптомы криза выражены и ацетон в моче более «++», ребенка госпитализируют и назначают лечение лекарственными препаратами: глюкоза, физиологический раствор, церукал и другие.
Однако борьба с ацетонемическим кризом и назначение некоторых лекарственных средств — это не основа лечения. Такой ребенок гораздо больше нуждается в охранном режиме и рациональном питании.
Можете ли вы помочь своему малышу?
Конечно! Ведь в этом случае от вас зависит гораздо больше, нежели от врача. Поэтому постарайтесь придерживаться некоторых советов:
* Оберегайте своего кроху от чрезмерных отрицательных эмоций и психических перегрузок, поскольку из-за эмоциональной лабильности малыш трудно их переносит.
* Ограничьте время нахождения ребенка за монитором компьютера и просмотра телепередач (особенно если он пристрастился к «стрелялкам»).
* Постепенно закаливайте малыша: утренняя зарядка, прогулки на свежем воздухе, влажные обтирания.
* Желательно, чтобы ребенок занимался физкультурой (но не интенсивным спортом!).
Кроме того, чтобы предотвратить избыточное поступление в организм пуринов, образование мочевой кислоты и кетоновых тел, важно придерживаться определенной диеты.
Диета
— Старайтесь, чтобы в рационе крохи в достаточном количестве присутствовали продукты, которые препятствуют образованию кетоновых тел в организме. В тоже время проследите, чтобы содержание пуриновых оснований и щавелевой кислоты в них было минимальным.
К ним относятся:
* молочные продукты (молоко, творог, сыр, сметана)
* большинство овощей и фруктов: картофель, кабачок, баклажан, яблоки, абрикосы и другие
* «защищенные» крупы (содержат крахмал): перловая, гречневая, овсяная
* мучные изделия
* яйца (не более одного в день).
— Постарайтесь, чтобы малыш получал в достаточном объеме щелочное питье (особенно во второй половине дня): минеральные воды, лимонную воду, свежеприготовленные фруктовые соки.
— Побалуйте своего кроху и добавьте немного «вкусненького»: мармелад, ягоды, сухофрукты (изюм, курага), желейные конфеты.
— Ограничьте (а в тяжелых случаях исключите) продукты, которые умеренно богаты пуриновыми основаниями:
нежирные сорта мяса (телятина, говядина, индюшатина, кролик, баранина)
морская рыба: макрель, камбала, пикша, семга, треска
раки
рыба речная: карп, судак, щука
лесные и грецкие орехи, миндаль
белый гриб, лисички, шампиньоны.
— Желательно, чтобы вы полностью исключили из рациона продукты, которые очень богаты пуринами и щавелевой кислотой, а также могут усилить ацидоз:
крепкие мясные и рыбные бульоны
курятина
субпродукты (мозги, почки, печень, язык)
продукты промышленного консервирования (если на них отсутствует пометка «Для детского питания»)
газированные напитки, в том числе любые другие напитки, которые долго хранились в холодильнике
бобовые (фасоль, соя)
шпинат, щавель, цветная капуста, петрушка, зеленый горошек, ревень
На заметку маме
- Сейчас уже не рекомендуется полностью исключать из рациона какао, чай, кофе и шоколад.
 Поскольку в результате проведенных исследований доказано, что из пуринов, содержащихся в них, не образуется мочевая кислота. Однако злоупотреблять ими все же не стоит, лучше ограничить.
Поскольку в результате проведенных исследований доказано, что из пуринов, содержащихся в них, не образуется мочевая кислота. Однако злоупотреблять ими все же не стоит, лучше ограничить. - Желательно, чтобы малыш употреблял все продукты в свежем, вареном и печеном виде.
 Поскольку в результате проведенных исследований доказано, что из пуринов, содержащихся в них, не образуется мочевая кислота. Однако злоупотреблять ими все же не стоит, лучше ограничить.
Поскольку в результате проведенных исследований доказано, что из пуринов, содержащихся в них, не образуется мочевая кислота. Однако злоупотреблять ими все же не стоит, лучше ограничить.
Конечно, ограничений достаточно много. Поэтому крохе сложно будет объяснить: почему, к примеру, он может лишь попробовать шоколад, тогда как соседский Мишка объедается этим лакомством.
Однако все же старайтесь придерживаться правильного питания, оберегайте своего малыша от чрезмерных эмоций и физических нагрузок. Ведь только благодаря этому ваш ребенок адаптируется к условиям внешней среды в разы быстрее и у него намного улучшится качество жизни. Ну, а вы сможете надолго забыть о запахе ацетона и привычных рвотах у вашего крохи.
фото: http://globallookpress.com/
Автор: Корецкая Валентина Петровна, педиатр,
врач-ординатор детского отделения
Нервно-артритический диатез — причины, симптомы, диагностика и лечение
Нервно-артритический диатез (НАД) – это конституционная аномалия, которая обусловлена нарушением метаболизма мочевой кислоты и пуринов. Проявляется неврастеническим, метаболическим, кожным и спастическим синдромами. Клиника зависит от присутствующего синдрома, симптоматически чаще всего наблюдаются гипертонус ЦНС, ацетонемическая рвота, расстройства стула. При диагностике НАД наибольшую ценность имеют анамнестические данные и результаты лабораторных исследований крови и мочи. Лечение включает в себя коррекцию рациона, психоэмоциональных и физических нагрузок. Проводится симптоматическая терапия, направленная на восстановление водно-электролитного баланса, по показаниям назначаются гепатопротекторы и витаминные препараты.
Проявляется неврастеническим, метаболическим, кожным и спастическим синдромами. Клиника зависит от присутствующего синдрома, симптоматически чаще всего наблюдаются гипертонус ЦНС, ацетонемическая рвота, расстройства стула. При диагностике НАД наибольшую ценность имеют анамнестические данные и результаты лабораторных исследований крови и мочи. Лечение включает в себя коррекцию рациона, психоэмоциональных и физических нагрузок. Проводится симптоматическая терапия, направленная на восстановление водно-электролитного баланса, по показаниям назначаются гепатопротекторы и витаминные препараты.
Общие сведения
Нервно-артритический или мочекислый диатез – это аномалия конституции, которая характеризуется склонностью к нарушению метаболизма мочевой кислоты и пуринов. Впервые данный термин ввел французский педиатр Жюль Комби в 1902 году. НАД – наименее распространенный вариант из всех форм диатезов, заболеваемость составляет 2-4% от общего количества детей. Лица мужского и женского пола страдают в равной степени, однако большинство характерных осложнений (ожирение, ацетонемическая рвота) чаще возникают у девочек. Клинические проявления зависят от возраста пациентов. В старшем возрасте нервно-артритический диатез нередко приводит к метаболическим заболеваниям, отложению солей в суставах, образованию камней в желчном пузыре и почках.
Лица мужского и женского пола страдают в равной степени, однако большинство характерных осложнений (ожирение, ацетонемическая рвота) чаще возникают у девочек. Клинические проявления зависят от возраста пациентов. В старшем возрасте нервно-артритический диатез нередко приводит к метаболическим заболеваниям, отложению солей в суставах, образованию камней в желчном пузыре и почках.
Нервно-артритический диатез
Причины нервно-артритического диатеза
Нервно-артритический диатез – это гетерогенная патология, которая обусловлена унаследованным нарушением обмена либо нерациональным питанием матери или ребенка. К наследственным факторам относятся дефекты генов, кодирующих метаболические энзимы пуринов и мочевой кислоты – фосфорибозилтрансферазы, фосфорибозилпирофосфатсинтетазы, глюкозо-6-фосфатазы и уратоксидазы. Как правило, семейный анамнез таких детей отягощен неврастенией, мочекаменной болезнью, невралгиями, подагрой или калькулезным холециститом. Алиментарным фактором, провоцирующим развитие НАД, является чрезмерное употребление белковой пищи матерью во время беременности или ребенком в раннем возрасте. Чаще всего это касается продуктов животного происхождения – говядины, телятины, птицы.
Чаще всего это касается продуктов животного происхождения – говядины, телятины, птицы.
В основе патогенеза нервно-артрического диатеза лежит гиперурикемия – повышение концентрации мочевой кислоты в плазме крови. Данное состояние влияет на ЦНС, стимулирует ее активность и восприимчивость к внешним раздражителям. Помимо прямого воздействия на ткани организма, гиперурикемия приводит к отложению солей в суставных оболочках, почках, желчном пузыре. Это явление основывается на кристаллизации уратов при попадании в суставную капсулу и взаимодействии с синовиальной жидкостью.
Симптомы нервно-артритического диатеза
Первые проявления нервно-артритического диатеза возникают уже в возрасте 1-2 месяца. Как правило, это неспецифические симптомы: снижение аппетита, нарушения сна, расстройства стула. Очень быстро развиваются дистрофические явления – медленная прибавка к массе тела или ожирение на фоне чрезмерного развития подкожной жировой клетчатки. Последний вариант чаще наблюдается у девочек. Также для НАД характерно умеренное увеличение всех групп лимфоузлов. Клиника нервно-артритического диатеза включает в себя следующие синдромы: неврастенический, метаболический, кожный, спастический. Редко данное патологическое состояние могут сопровождать субфебрильная температура, резкое отвращение к запахам.
Последний вариант чаще наблюдается у девочек. Также для НАД характерно умеренное увеличение всех групп лимфоузлов. Клиника нервно-артритического диатеза включает в себя следующие синдромы: неврастенический, метаболический, кожный, спастический. Редко данное патологическое состояние могут сопровождать субфебрильная температура, резкое отвращение к запахам.
Неврастенический синдром наиболее распространен и присутствует у 85% детей с нервно-артритическим диатезом. В младшем возрасте он проявляется чрезмерной пугливостью, беспокойством, бессонницей, тревожным и поверхностным сном. На фоне гиперурикемии происходит преждевременное развитие ЦНС, из-за чего дети рано начинают говорить и читать, очень любознательны, обладают хорошей зрительной и словесной памятью. Кроме этого, отмечается эмоциональная нестабильность, могут наблюдаться ночные кошмары, потеря аппетита. Часто при нервно-артритическом диатезе развиваются вегето-сосудистая дистония, нервные тики, психогенный кашель и рвота, энурез, логоневроз, аэрофагия.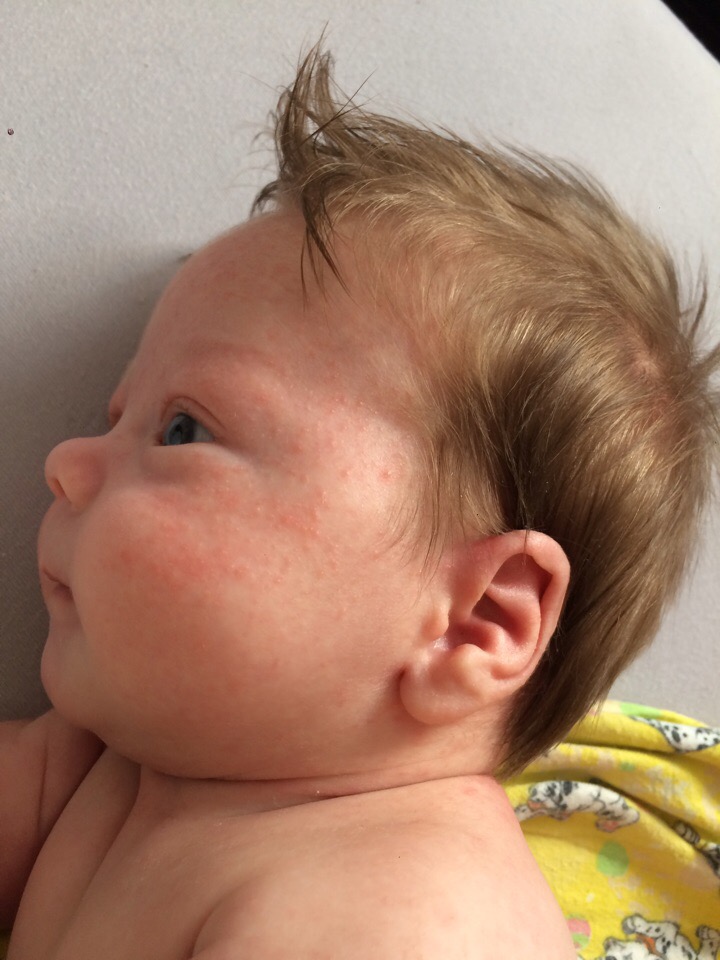
Метаболический синдром включает в себя периодические боли в суставах, которые обостряются в ночное время, перемежающиеся нарушения мочеиспускания, ацетонемическую рвоту. Данные симптомы характерны для детей в возрасте 2 лет. Боли возникают преимущественно в мелких суставах дистальных отделов пальцев рук и ног. Дизурические явления связаны со сниженной концентрационной способностью почек и отложением в них солей. Клинически это может проявляться тупой болью в пояснице, потемнением мочи. Ацетонемический синдром при нервно-артритическом диатезе более характерен для девочек и исчезает при переходе в пубертатный период. Приступ возникает остро, как правило, ему предшествует стресс, физическое перенапряжение, несоблюдение диеты. Рвота неукротима, может сопровождаться эксикозом, редко – судорогами. Длительность приступа колеблется от 1-2 до 24-36 часов.
Аллергическо-дерматический синдром. Кожная сыпь при нервно-артритическом диатезе возникает довольно редко, но при ее наличии может наблюдаться зуд. В старшем возрасте возможно формирование крапивницы, нейродермита, сухой или себорейной экземы. У некоторых детей на фоне ОРВИ наблюдается навязчивый кашель, чихание. Иногда на фоне НАД развивается астматический бронхит, который может усугубляться спастическим синдромом и переходить в атопическую бронхиальную астму.
В старшем возрасте возможно формирование крапивницы, нейродермита, сухой или себорейной экземы. У некоторых детей на фоне ОРВИ наблюдается навязчивый кашель, чихание. Иногда на фоне НАД развивается астматический бронхит, который может усугубляться спастическим синдромом и переходить в атопическую бронхиальную астму.
Спастический синдром. Для детей с нервно-артритическим диатезом характерна склонность к спазмированию гладкомышечной мускулатуры стенок бронхов, кишечника и сосудов. Это может проявляться обструкцией бронхиального дерева, кишечными, почечными и печеночными коликами, мембранозным колитом, запорами, мигренью и гипертонией.
Диагностика нервно-артритического диатеза
Диагностика нервно-артритического диатеза основывается на сборе анамнеза, объективном осмотре, результатах лабораторных и инструментальных тестов. Анамнестические данные позволяют установить наличие нарушения метаболизма пуринов и мочевой кислоты у родителей, братьев и сестер. Также выясняются темпы психического развития и ранние симптомы НАД. При физикальном обследовании педиатром могут определяться признаки гипертонуса нервной системы, запах ацетона изо рта, дефицит массы тела, редко – кожная сыпь.
Также выясняются темпы психического развития и ранние симптомы НАД. При физикальном обследовании педиатром могут определяться признаки гипертонуса нервной системы, запах ацетона изо рта, дефицит массы тела, редко – кожная сыпь.
Ведущую роль в диагностике нервно-артритического диатеза играют лабораторные анализы. При ацетонемическом кризе в крови ребенка выявляются гиперурикемия, лейкоцитоз со сдвигом формулы влево, эозинопения, моноцитопения и лимфоцитопения, гипогликемия, кетонемия, метаболический ацидоз, повышение СОЭ. В моче определяются протеинурия, микрогематурия, наличие солей уратов, фосфатов и оксалатов. В состоянии компенсации перечисленные отклонения от нормы минимальны или отсутствуют. В зависимости от клинической формы нервно-артритического диатеза могут присутствовать другие изменения в анализах или клинические симптомы. Инструментальная диагностика в виде УЗИ, рентгенографии и иных методов применяется для исключения органических патологий и проведения дифференциального диагноза.
Дифференциальная диагностика нервно-артритического диатеза в педиатрии проводится с такими заболеваниями, как ревматизм, неврозы разнообразного генеза, пиелонефрит, панкреатит, холецистит, синдромом Леша-Нихена, синдром минимальной мозговой дисфункции, туберкулезная интоксикация.
Лечение нервно-артритического диатеза
Основу лечения у детей с нервно-артритическим диатезом составляет правильное питание, ограничение физического и психоэмоционального напряжения. Основу рациона ребенка должны составлять свежие фрукты и овощи, молоко и кисломолочные продукты, каши из гречневой, овсяной, перловой, пшеничной крупы. Строго ограничивается употребление мяса, рыбы, копченостей, жареной и жирной пищи, кондитерских изделий. Детям с нервно-артритическим диатезом категорически противопоказаны продукты, которые содержат большое количество пуринов и кофеина – шоколад, какао, кофе, паштет и т. д. Важно соблюдение режима, прием пищи должен проходить 4-5 раз в сутки в одно и то же время.
При развитии ацетонемического криза ребенку следует давать подслащенный чай, щелочные минеральные воды, свежие соки, физраствор, 5% глюкозу с частотой каждые 10-15 минут. Также показана очистительная клизма. В некоторых случаях применяются гепатопротекторы и витамины группы В. При ацетонемической рвоте проводится коррекция метаболического ацидоза и дегидратации. При низкой интенсивности данных осложнений показана оральная регидратация вышеуказанными жидкостями, в тяжелых случаях – внутривенная инфузия физраствора, глюкозы, витамина С, гепатопротекторов. Предварительно осуществляется промывание желудка и кишечника.
Прогноз и профилактика нервно-артритического диатеза
Прогноз для жизни при нервно-артритическом диатезе благоприятный, для здоровья – сомнительный. У таких людей рано возникают артериальная гипертензия, подагра, атеросклероз, метаболические артриты, сахарный диабет, часто формируются желчнокаменная и мочекаменная болезни, ожирение. При подтверждении НАД пациент должен регулярно посещать лечащего педиатра или семейного врача с целью ранней диагностики и своевременного лечения перечисленных заболеваний. Специфической профилактики нервно-артритического диатеза не существует. Для предотвращения кризов следует соблюдать диету, избегать чрезмерных физических и психических нагрузок. Антенатальная профилактика заключается в медико-генетическом консультировании семейных пар, в особенности при наличии патологий, связанных с нарушением метаболизма.
При подтверждении НАД пациент должен регулярно посещать лечащего педиатра или семейного врача с целью ранней диагностики и своевременного лечения перечисленных заболеваний. Специфической профилактики нервно-артритического диатеза не существует. Для предотвращения кризов следует соблюдать диету, избегать чрезмерных физических и психических нагрузок. Антенатальная профилактика заключается в медико-генетическом консультировании семейных пар, в особенности при наличии патологий, связанных с нарушением метаболизма.
Нервно-артритический диатез — причины, симптомы, диагностика и лечение
Нервно-артритический диатез (НАД) – это конституционная аномалия, которая обусловлена нарушением метаболизма мочевой кислоты и пуринов. Проявляется неврастеническим, метаболическим, кожным и спастическим синдромами. Клиника зависит от присутствующего синдрома, симптоматически чаще всего наблюдаются гипертонус ЦНС, ацетонемическая рвота, расстройства стула. При диагностике НАД наибольшую ценность имеют анамнестические данные и результаты лабораторных исследований крови и мочи. Лечение включает в себя коррекцию рациона, психоэмоциональных и физических нагрузок. Проводится симптоматическая терапия, направленная на восстановление водно-электролитного баланса, по показаниям назначаются гепатопротекторы и витаминные препараты.
При диагностике НАД наибольшую ценность имеют анамнестические данные и результаты лабораторных исследований крови и мочи. Лечение включает в себя коррекцию рациона, психоэмоциональных и физических нагрузок. Проводится симптоматическая терапия, направленная на восстановление водно-электролитного баланса, по показаниям назначаются гепатопротекторы и витаминные препараты.
Общие сведения
Нервно-артритический или мочекислый диатез – это аномалия конституции, которая характеризуется склонностью к нарушению метаболизма мочевой кислоты и пуринов. Впервые данный термин ввел французский педиатр Жюль Комби в 1902 году. НАД – наименее распространенный вариант из всех форм диатезов, заболеваемость составляет 2-4% от общего количества детей. Лица мужского и женского пола страдают в равной степени, однако большинство характерных осложнений (ожирение, ацетонемическая рвота) чаще возникают у девочек. Клинические проявления зависят от возраста пациентов.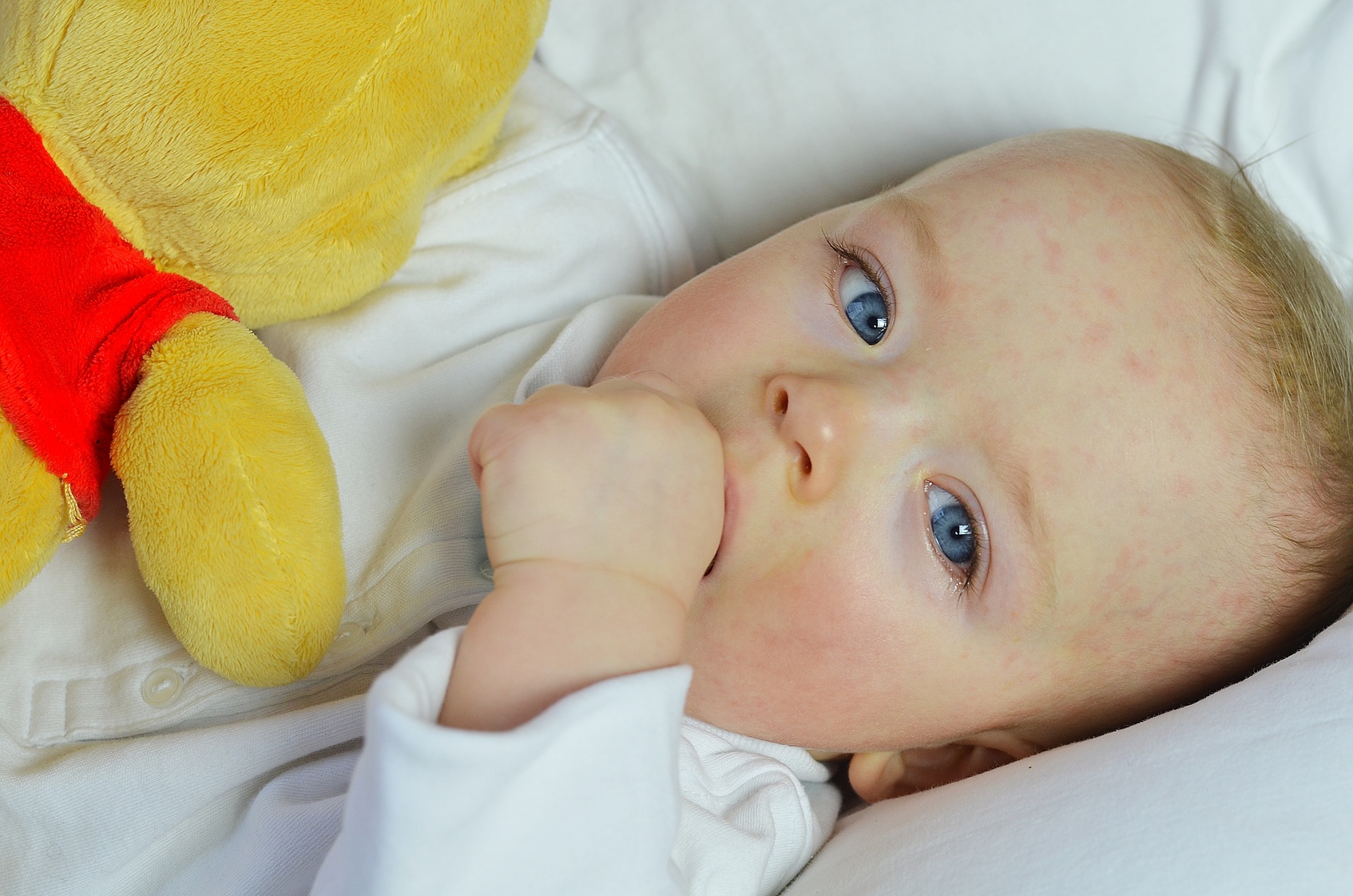 В старшем возрасте нервно-артритический диатез нередко приводит к метаболическим заболеваниям, отложению солей в суставах, образованию камней в желчном пузыре и почках.
В старшем возрасте нервно-артритический диатез нередко приводит к метаболическим заболеваниям, отложению солей в суставах, образованию камней в желчном пузыре и почках.
Нервно-артритический диатез
Причины нервно-артритического диатеза
Нервно-артритический диатез – это гетерогенная патология, которая обусловлена унаследованным нарушением обмена либо нерациональным питанием матери или ребенка. К наследственным факторам относятся дефекты генов, кодирующих метаболические энзимы пуринов и мочевой кислоты – фосфорибозилтрансферазы, фосфорибозилпирофосфатсинтетазы, глюкозо-6-фосфатазы и уратоксидазы. Как правило, семейный анамнез таких детей отягощен неврастенией, мочекаменной болезнью, невралгиями, подагрой или калькулезным холециститом. Алиментарным фактором, провоцирующим развитие НАД, является чрезмерное употребление белковой пищи матерью во время беременности или ребенком в раннем возрасте. Чаще всего это касается продуктов животного происхождения – говядины, телятины, птицы.
В основе патогенеза нервно-артрического диатеза лежит гиперурикемия – повышение концентрации мочевой кислоты в плазме крови. Данное состояние влияет на ЦНС, стимулирует ее активность и восприимчивость к внешним раздражителям. Помимо прямого воздействия на ткани организма, гиперурикемия приводит к отложению солей в суставных оболочках, почках, желчном пузыре. Это явление основывается на кристаллизации уратов при попадании в суставную капсулу и взаимодействии с синовиальной жидкостью.
Симптомы нервно-артритического диатеза
Первые проявления нервно-артритического диатеза возникают уже в возрасте 1-2 месяца. Как правило, это неспецифические симптомы: снижение аппетита, нарушения сна, расстройства стула. Очень быстро развиваются дистрофические явления – медленная прибавка к массе тела или ожирение на фоне чрезмерного развития подкожной жировой клетчатки. Последний вариант чаще наблюдается у девочек. Также для НАД характерно умеренное увеличение всех групп лимфоузлов. Клиника нервно-артритического диатеза включает в себя следующие синдромы: неврастенический, метаболический, кожный, спастический. Редко данное патологическое состояние могут сопровождать субфебрильная температура, резкое отвращение к запахам.
Клиника нервно-артритического диатеза включает в себя следующие синдромы: неврастенический, метаболический, кожный, спастический. Редко данное патологическое состояние могут сопровождать субфебрильная температура, резкое отвращение к запахам.
Неврастенический синдром наиболее распространен и присутствует у 85% детей с нервно-артритическим диатезом. В младшем возрасте он проявляется чрезмерной пугливостью, беспокойством, бессонницей, тревожным и поверхностным сном. На фоне гиперурикемии происходит преждевременное развитие ЦНС, из-за чего дети рано начинают говорить и читать, очень любознательны, обладают хорошей зрительной и словесной памятью. Кроме этого, отмечается эмоциональная нестабильность, могут наблюдаться ночные кошмары, потеря аппетита. Часто при нервно-артритическом диатезе развиваются вегето-сосудистая дистония, нервные тики, психогенный кашель и рвота, энурез, логоневроз, аэрофагия.
Метаболический синдром включает в себя периодические боли в суставах, которые обостряются в ночное время, перемежающиеся нарушения мочеиспускания, ацетонемическую рвоту. Данные симптомы характерны для детей в возрасте 2 лет. Боли возникают преимущественно в мелких суставах дистальных отделов пальцев рук и ног. Дизурические явления связаны со сниженной концентрационной способностью почек и отложением в них солей. Клинически это может проявляться тупой болью в пояснице, потемнением мочи. Ацетонемический синдром при нервно-артритическом диатезе более характерен для девочек и исчезает при переходе в пубертатный период. Приступ возникает остро, как правило, ему предшествует стресс, физическое перенапряжение, несоблюдение диеты. Рвота неукротима, может сопровождаться эксикозом, редко – судорогами. Длительность приступа колеблется от 1-2 до 24-36 часов.
Данные симптомы характерны для детей в возрасте 2 лет. Боли возникают преимущественно в мелких суставах дистальных отделов пальцев рук и ног. Дизурические явления связаны со сниженной концентрационной способностью почек и отложением в них солей. Клинически это может проявляться тупой болью в пояснице, потемнением мочи. Ацетонемический синдром при нервно-артритическом диатезе более характерен для девочек и исчезает при переходе в пубертатный период. Приступ возникает остро, как правило, ему предшествует стресс, физическое перенапряжение, несоблюдение диеты. Рвота неукротима, может сопровождаться эксикозом, редко – судорогами. Длительность приступа колеблется от 1-2 до 24-36 часов.
Аллергическо-дерматический синдром. Кожная сыпь при нервно-артритическом диатезе возникает довольно редко, но при ее наличии может наблюдаться зуд. В старшем возрасте возможно формирование крапивницы, нейродермита, сухой или себорейной экземы. У некоторых детей на фоне ОРВИ наблюдается навязчивый кашель, чихание. Иногда на фоне НАД развивается астматический бронхит, который может усугубляться спастическим синдромом и переходить в атопическую бронхиальную астму.
Иногда на фоне НАД развивается астматический бронхит, который может усугубляться спастическим синдромом и переходить в атопическую бронхиальную астму.
Спастический синдром. Для детей с нервно-артритическим диатезом характерна склонность к спазмированию гладкомышечной мускулатуры стенок бронхов, кишечника и сосудов. Это может проявляться обструкцией бронхиального дерева, кишечными, почечными и печеночными коликами, мембранозным колитом, запорами, мигренью и гипертонией.
Диагностика нервно-артритического диатеза
Диагностика нервно-артритического диатеза основывается на сборе анамнеза, объективном осмотре, результатах лабораторных и инструментальных тестов. Анамнестические данные позволяют установить наличие нарушения метаболизма пуринов и мочевой кислоты у родителей, братьев и сестер. Также выясняются темпы психического развития и ранние симптомы НАД. При физикальном обследовании педиатром могут определяться признаки гипертонуса нервной системы, запах ацетона изо рта, дефицит массы тела, редко – кожная сыпь.
Ведущую роль в диагностике нервно-артритического диатеза играют лабораторные анализы. При ацетонемическом кризе в крови ребенка выявляются гиперурикемия, лейкоцитоз со сдвигом формулы влево, эозинопения, моноцитопения и лимфоцитопения, гипогликемия, кетонемия, метаболический ацидоз, повышение СОЭ. В моче определяются протеинурия, микрогематурия, наличие солей уратов, фосфатов и оксалатов. В состоянии компенсации перечисленные отклонения от нормы минимальны или отсутствуют. В зависимости от клинической формы нервно-артритического диатеза могут присутствовать другие изменения в анализах или клинические симптомы. Инструментальная диагностика в виде УЗИ, рентгенографии и иных методов применяется для исключения органических патологий и проведения дифференциального диагноза.
Дифференциальная диагностика нервно-артритического диатеза в педиатрии проводится с такими заболеваниями, как ревматизм, неврозы разнообразного генеза, пиелонефрит, панкреатит, холецистит, синдромом Леша-Нихена, синдром минимальной мозговой дисфункции, туберкулезная интоксикация.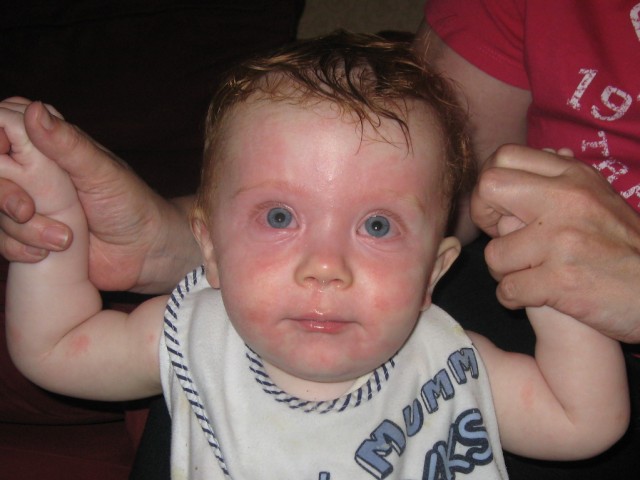
Лечение нервно-артритического диатеза
Основу лечения у детей с нервно-артритическим диатезом составляет правильное питание, ограничение физического и психоэмоционального напряжения. Основу рациона ребенка должны составлять свежие фрукты и овощи, молоко и кисломолочные продукты, каши из гречневой, овсяной, перловой, пшеничной крупы. Строго ограничивается употребление мяса, рыбы, копченостей, жареной и жирной пищи, кондитерских изделий. Детям с нервно-артритическим диатезом категорически противопоказаны продукты, которые содержат большое количество пуринов и кофеина – шоколад, какао, кофе, паштет и т. д. Важно соблюдение режима, прием пищи должен проходить 4-5 раз в сутки в одно и то же время.
При развитии ацетонемического криза ребенку следует давать подслащенный чай, щелочные минеральные воды, свежие соки, физраствор, 5% глюкозу с частотой каждые 10-15 минут. Также показана очистительная клизма. В некоторых случаях применяются гепатопротекторы и витамины группы В. При ацетонемической рвоте проводится коррекция метаболического ацидоза и дегидратации. При низкой интенсивности данных осложнений показана оральная регидратация вышеуказанными жидкостями, в тяжелых случаях – внутривенная инфузия физраствора, глюкозы, витамина С, гепатопротекторов. Предварительно осуществляется промывание желудка и кишечника.
При ацетонемической рвоте проводится коррекция метаболического ацидоза и дегидратации. При низкой интенсивности данных осложнений показана оральная регидратация вышеуказанными жидкостями, в тяжелых случаях – внутривенная инфузия физраствора, глюкозы, витамина С, гепатопротекторов. Предварительно осуществляется промывание желудка и кишечника.
Прогноз и профилактика нервно-артритического диатеза
Прогноз для жизни при нервно-артритическом диатезе благоприятный, для здоровья – сомнительный. У таких людей рано возникают артериальная гипертензия, подагра, атеросклероз, метаболические артриты, сахарный диабет, часто формируются желчнокаменная и мочекаменная болезни, ожирение. При подтверждении НАД пациент должен регулярно посещать лечащего педиатра или семейного врача с целью ранней диагностики и своевременного лечения перечисленных заболеваний. Специфической профилактики нервно-артритического диатеза не существует. Для предотвращения кризов следует соблюдать диету, избегать чрезмерных физических и психических нагрузок. Антенатальная профилактика заключается в медико-генетическом консультировании семейных пар, в особенности при наличии патологий, связанных с нарушением метаболизма.
Антенатальная профилактика заключается в медико-генетическом консультировании семейных пар, в особенности при наличии патологий, связанных с нарушением метаболизма.
Нервно-артритический диатез у детей: симптомы, лечение, характерные проявления
Нервно-артритический диатез у детей – патология, вызванная нарушением обмена мочевой кислоты и пуринов. Характеризуется избытком аммиачных соединений в организме, на фоне которого возникает повышенная нервная возбудимость и плохой аппетит. Заболевание поддается лечению, главным образом через организацию диетического питания.
Нервно артритический диатез у детей может быть врождённым или приобретенным
Особенности нервно-артритического диатеза у детей
Патология имеет несколько форм проявления: спастический, неврастенический, кожный или метаболический синдром. Клиническая картина заболевания зависит от его формы, в большинстве случаев сопровождается повышенным тонусом центральной нервной системы, расстройством работы кишечника, рвотой.
Нервно-артритическая форма диатеза составляет не более 4% от всех форм детских диатезов. Заболеванию в равной степени подвержены оба пола, но осложнения диагностируются чаще всего у девочек.
Причины патологии
Причиной развития заболевания может быть унаследованная патология обмена веществ или перекос в питании матери во время беременности или ребенка в первые годы жизни.
Генные поломки, провоцирующие нарушение метаболизма мочевой кислоты и пуринов, отягощают семейный анамнез ребенка такими заболеваниями:
- неврастения;
- калькулезный холецистит;
- подагра;
- мочекаменная болезнь;
- невралгия.
Приобретенная форма диатеза проявляется вследствие преобладания в рационе матери или ребенка белковой пищи животного происхождения.
В зарождении и развитии нервно-артритического диатеза основным фактором является гиперурикемия – состояние, при котором в крови отмечается повышенное содержание мочевой кислоты. В результате оказывается негативное влияние на работу нервной системы, возникают отложения кристаллизированных уратов в капсулах суставов и органах мочевыводящей системы.
В результате оказывается негативное влияние на работу нервной системы, возникают отложения кристаллизированных уратов в капсулах суставов и органах мочевыводящей системы.
Симптомы заболевания
Симптомы заболевания зависят от его клинической формы
Первая симптоматика НАД проявляется уже на втором месяце жизни младенца. Ребенок может плохо кушать и спать, появляется расстройство стула. По мере роста появляются пролемы с весом — дистрофия или, напротив, ожирение. Чрезмерные отложения жира фиксируются преимущественно у девочек. Характерное проявление НАД — умеренно гипотрофированные лимфатические узлы всех групп.
Перечень симптомов в зависимости от синдрома НАД:
1. Неврастенический синдром. Самый распространенный, диагностируется в 85% случаев. Характерные проявления:
- нарушение сна;
- лабильность нервной системы и эмоциональной сферы;
- тревожность, пугливость, беспокойство;
- опережающее возрастную норму развитие центральной нервной системы;
- плохой аппетит;
- вегето-сосудистая дистония;
- логоневроз;
- тики;
- энурез;
- аэрофагия;
- психогенная рвота и кашель.

Такие дети могут начать говорить и читать раньше сверстников, активно познавать мир, демонстрировать хорошую память, внимание.
2. Метаболический синдром. Характеризуется наличием следующих симптомов, сменяющих друг друга:
- болезненность мелких суставов рук и ног, нарастающая ночью;
- дизурия, сопровождающаяся тупой болью в поясничном отделе;
- резкий приступ неукротимой ацетонемической рвоты на фоне стресса, погрешности в питании или перенапряжения, которая может приводить к обезвоживанию организма, реже — судорогам.
Продолжительность такого приступа может достигать 36 часов.
3. Аллергическо-дерматический синдром. Характеризуется появлением зудящей сыпи на коже. Могут присоединиться такие симптомы:
- экзема;
- крапивница;
- астматический бронхит;
- нейродермит;
- навязчивое чихание и кашель на фоне острой респираторной вирусной инфекции
4. Спастический синдром. Основные признаки:
Спастический синдром. Основные признаки:
- бронхообструкция;
- бронхиолит;
- колиты;
- колики;
- запор;
- мигрень;
- гипертония.
К общим признакам патологии можно отнести субфебрилитет и гиперосмию.
Диагностика патологического состояния
Обследование пациента включает в себя сбор семейного анамнеза, в ходе которого устанавливается наличие расстройств метаболизма у ближайших родственников и ранних симптомов нервно-артрического диатеза у ребенка. Осмотр педиатра позволяет выявить признаки повышенного тонуса, несоответствие фактического веса возрастным нормам, наличие кожных высыпаний и запаха ацетона из ротовой полости.
Проведение лабораторных и инструментальных исследований играет основную роль в постановке диагноза. В анализе крови отмечается изменение клеточного состава, повышение скорости оседания эритроцитов, снижение числа эозинофилов, моноцитов, лимфоцитов и концентрации глюкозы, метаболический ацидоз, ацетонемия.
Чем выше компенсаторные возможности организма пациента, тем меньшее отклонение от нормы будут иметь анализы крови.
Анализ мочи при нервно-артритическом диатезе показывает наличие белка и солей уратов, высокое содержание эритроцитов.
В зависимости от формы НАД к перечисленным отклонениям анализов могут добавляться иные перекосы в составе крови и мочи.
Дифференциальная диагностика патологии проводится методами ультразвукового исследования и рентгенографии. Исключать следует такие заболевания:
- спазмофилия;
- невроз любого генеза;
- ревматизм;
- синдром ММД;
- туберкулез;
- холецистит;
- пиелонефрит;
- синдром Леша-Нихена;
- панкреатит.
Для диагностики и терапии заболевания следует обратиться к врачу-педиатру.
Лечение НАД
Лечение заключается в диете и снижении психоэмоциональной и физической нагрузки
В основе терапии заболевания лежит снижение психоэмоциональной и физической нагрузки и организация правильного диетического питания.
Купирование ацетонемического криза проводят отпаиванием чистой питьевой или минеральной водой, солевыми растворами, 5% глюкозой, препаратами, восстанавливающими печент, витаминами группы В.
Перед проведением оральной регидратации или, в тяжелых случаях пациенту показана очистительная клизма.
При ацетонемической рвоте требуется коррекция кислотно-щелочного баланса в организме и профилактика обезвоживания.
Диета
Низкобелковая диета ребенка должна состоять преимущественно из свежих овощей и фруктов, молочных и кисломолочных продуктов, круп. В рационе питания должно быть минимум мясных, рыбных, жареных, копченых, жирных блюд, бульонов, холодца и кондитерских изделий.
Во время лечения следует полностью исключить из рациона малыша кофеин, крепкий чай, шоколадные и содержащие какао продукты, соленья, пряности, колбасы, грибы, консервы, зеленый горошек, щавель, цветную капусту.
В вечернее время ребенка следует кормить гречей, овсянкой, картофелем, овощами.
Немаловажным при соблюдении диеты является режим питания. Следует организовать 5 приемов пищи в день, в одно и то же время каждый день.
Профилактические мероприятия и прогноз
Женщина на этапах планирования и вынашивания беременности должна уделять внимание своему рациону, избегать нарушений в питании, потреблять больше свежих овощей и фруктов, кисломолочных продуктов.
Основой профилактики НАД у детей является соблюдение рационального режима дня, включающее в себя ежедневные прогулки на свежем воздухе, полноценный сон не менее 9-11 часов в зависимости от возраста, физические упражнения, закаливающие процедуры.
Рекомендуется избегать сезонных авитаминозов, насыщать организм ребенка всеми необходимыми витаминами и минералами.
Не менее важен эмоциональный настрой малыша. Следует исключить любые психотравмирующие ситуации, стрессы, конфликты, психологические нагрузки, ребенок должен расти в спокойной, доброжелательной обстановке.
Следует исключить любые психотравмирующие ситуации, стрессы, конфликты, психологические нагрузки, ребенок должен расти в спокойной, доброжелательной обстановке.
При своевременном обращении к специалистам и соблюдении всех предписаний прогноз для дальнейшей жизни благоприятный, но полностью избавиться от недуга невозможно. Кроме того на его фоне нередко возникают такие заболевания:
- подагра;
- ожирение;
- болезни мочевыводящей системы;
- артериальная гипертензия;
- сахарный диабет;
- атеросклероз;
- артрит.
Пациент с диагностированным нервно-артрическим диатезом обязательно должен находиться на контроле врача, своевременно сдавать анализы и проходить обследования. Чем раньше будут обнаружены признаки сопутствующих заболеваний, тем выше шансы на их купирование и лечение.
Семьи, в анамнезе которых имеются патологии обмена веществ, обязательно должны посетить генетика перед планированием беременности. Только профилактика и своевременная диагностика состояния способны предотвратить развитие недуга и его опасных осложнений.
Только профилактика и своевременная диагностика состояния способны предотвратить развитие недуга и его опасных осложнений.
Читайте далее: причины развития аллергического диатеза у детей
фото и лечение экссудативного, и других диатезов у детей
К сожалению, даже у тех новорожденных, что находятся на грудном вскармливании, зачастую проявляются аллергические реакции. Что уж говорить о тех крохах, которые получают искусственное питание! Самыми распространенными видами диатезов у детей являются экссудативный (атопический дерматит), нервно-артритический и лимфатико-гипопластический. При каждом из них показано свое лечение.
Атопический дерматит у детей: фото, причины и симптомы
Говоря о том, какие бывают диатезы у детей, в первую очередь стоит описать экссудативный (атопический дерматит). Проявления экссудативного диатеза в виде поражения кожи и слизистых оболочек той или иной степени имеют более половины малышей первого года жизни. В настоящее время термин «экссудативный диатез» у детей заменен на новое название — атопический дерматит, но суть заболевания от переименования не изменилась.
В настоящее время термин «экссудативный диатез» у детей заменен на новое название — атопический дерматит, но суть заболевания от переименования не изменилась.
Главной причиной развития атопического дерматита у детей является аллергия. У одних детей диатез носит непостоянный и кратковременный характер, а у других растет вместе с ними, трансформируясь в будущем в многочисленные аллергические заболевания.
Больше всего страдают от атопического дерматита дети-«искусственники». Объясняется все довольно просто. Барьерная функция кишечника и печени у младенцев значительно снижена в силу анатомо-физиологических особенностей. Поэтому кишечная стенка является весьма проницаемой для не успевших расщепиться крупных молекул, играющих роль аллергенов, и беспрепятственно пропускает их в кровеносное русло. А печень со слабо выраженной обезвреживающей способностью не способна справиться с агрессивными молекулами, и они спокойно циркулируют в организме, вызывая аллергические реакции.
Перечень аллергенных продуктов можно продолжать до бесконечности, так как у каждого конкретного ребенка имеются «свои» конкретные «недруги». Но наиболее опасными продуктами, вызывающими клинические проявления экссудативного диатеза у большинства детей, являются коровье молоко, яйца, шоколад, цитрусовые, орехи, клубника, рыба. Дети, получающие грудь матери, надежно защищены от диатеза, но в некоторых случаях проявления диатеза появляются у них при употреблении аллергенных продуктов матерью.
Но наиболее опасными продуктами, вызывающими клинические проявления экссудативного диатеза у большинства детей, являются коровье молоко, яйца, шоколад, цитрусовые, орехи, клубника, рыба. Дети, получающие грудь матери, надежно защищены от диатеза, но в некоторых случаях проявления диатеза появляются у них при употреблении аллергенных продуктов матерью.
Симптомы атопического дерматита у детей могут проявиться уже в первые дни после рождения в виде желтоватых чешуек на голове и опрелостей в кожных складках.
Как видно на фото, экссудативный диатез у детей чаще всего проявляется в виде покраснения и шелушения щечек (так называемый «молочный струп»):
При атопическом дерматите грудной ребенок довольно спокойно переносит диатезный «румянец», но иногда ему досаждают зудящие щечки, и он способен расчесывать их. Правильно налаженное питание способно справиться с диатезом, и кожа полностью очищается. Возможны обострения процесса, возникающие при нарушении диеты, под влиянием профилактической прививки, при развитии дисбактериоза, при любом заболевании.
Сильно выраженный процесс становится настоящей бедой для малыша и всей семьи.
Посмотрите на фото – при атопическом дерматите у детей на коже появляются обширные покрасневшие участки с множественными пузырьками, которые лопаются и сливаются между собой, образуя мокнущие поверхности, покрывающиеся корочками, нередко с присоединением вторичной гнойной инфекции:
Ребенок становится беспокойным и раздражительным, а постоянный кожный зуд делает сон поверхностным и прерывистым. Страдает аппетит: иногда понижается, но чаще повышается, и ребенок становится полным, рыхлым, а в жировых складочках проявления диатеза находят для себя наиболее благоприятные условия. Часто наблюдается неустойчивый стул, связанный с изменениями на слизистой желудочно-кишечного тракта, ферментативной недостаточностью и нарушением всасываемости пищевых веществ.
Посмотреть, как выглядят проявления этого вида диатеза у детей можно на фото, представленных ниже:
Дети с экссудативным диатезом предрасположены к респираторным инфекциям, нередко с обструктивным синдромом (спазм бронхов), стенозам гортани (ложный круп), конъюнктивитам, дисбактериозу, кишечным инфекциям.
В последние годы во всем мире наблюдается необычайный всплеск аллергических заболеваний, в том числе и проявлений экссудативного диатеза у детей первого года жизни. Поэтому врачи настойчиво призывают молодых женщин озаботиться профилактикой экссудативного диатеза у ребенка еще во время беременности и исключить из своего питания аллергизирующие продукты. Эти же требования распространяется на кормящую маму, а «искусственнику» следует подобрать адаптированную смесь, которая не вызывает у него аллергию. Легко сказать «подобрать». На самом деле процесс этот длительный, нервный и дорогостоящий, так как каждая пачка смеси после неудачной пробной порции становится ненужной, подрывая семейный бюджет и вынуждая к приобретению следующей.
Как лечить атопический дерматит у грудного ребенка: препараты для лечения детей
Лечение атопического дерматита у детей заключается не только в диетотерапии.
Очень важно вводить в организм достаточное количество витаминов:
- витамин В6 — по 50-75 мг в сутки;
- витамин А — 5-7-10 тысяч ME в сутки в течение 3 недель;
- витамин Е — 25-30 мг в сутки;
- пантотенат кальция (витамин В5) — 100 мг в сутки;
- пангамат кальция (витамин В15) — по 50-100 мг в сутки.

Эффективными препаратами для лечения атопического дерматита у детей при обострении процесса из-за явного нарушения диеты являются адсорбенты для связывания аллергенов в желудочно-кишечном тракте и удаления их из организма: активированный уголь, смекта, полифепан, полисорб, энтеросгель.
Перед тем как лечить атопический дерматит у ребенка, обязательно посоветуйтесь с аллергологом. Известно, что экссудативный диатез и пищевая аллергия идут рука об руку с дисбактериозом. Поэтому в комплексное лечение атопического дерматита детей входят препараты, улучшающие состояние кишечной микрофлоры: лактобактерин, бифидумбактерин, эуфлорин, линекс и т. д. курсами по 10 дней.
Большое значение имеет применение антигистаминных препаратов (супрастин, тавегил, кларитин, фенистил, зиртек), курсами по 7-10 дней и микроэлементов (кальций, фосфор, цинк, магний).
Как помочь ребенку при атопическом дерматите: лучшие средства лечения
В повышенном внимании и тщательном уходе нуждается поврежденная кожа. Чтобы помочь ребенку при атопическом дерматите, одежда должна быть только из хлопчатобумажных тканей. Не кутайте ребенка! Повышенная потливость усиливает зуд и приводит к обострению процесса. Избегайте пользоваться мылом и моющими средствами, обезжиривающими кожу. При купании ребенка используйте травяные отвары (череда, чистотел, ромашка). Белье стирайте только детским мылом и тщательно прополаскивайте. У многих детей проявления экссудативного диатеза усиливаются при использовании одноразовых подгузников. Придется отказаться от них и перейти на марлевые. Но и клеенка, подложенная под простынку, способствует обострению процесса. В таком случае используйте одноразовые хлопчатобумажные пеленки с повышенной впитывающей способностью. Они уберегут кроватку и мягкую мебель от «наводнения».
Чтобы помочь ребенку при атопическом дерматите, одежда должна быть только из хлопчатобумажных тканей. Не кутайте ребенка! Повышенная потливость усиливает зуд и приводит к обострению процесса. Избегайте пользоваться мылом и моющими средствами, обезжиривающими кожу. При купании ребенка используйте травяные отвары (череда, чистотел, ромашка). Белье стирайте только детским мылом и тщательно прополаскивайте. У многих детей проявления экссудативного диатеза усиливаются при использовании одноразовых подгузников. Придется отказаться от них и перейти на марлевые. Но и клеенка, подложенная под простынку, способствует обострению процесса. В таком случае используйте одноразовые хлопчатобумажные пеленки с повышенной впитывающей способностью. Они уберегут кроватку и мягкую мебель от «наводнения».
Хорошее лечебное воздействие оказывает пребывание на ласковом солнышке (не злоупотреблять!) и купание в морской воде (можно в ванне с добавлением морской соли).
При обширных мокнущих участках применяются примочки с 2% раствором танина, 0,25% раствором сульфата цинка, 5% раствором жидкости Бурова, отварами подорожника, ромашки. Очень хорошо зарекомендовало себя такое средство лечения атопического дерматита у детей, как лосьон «Каламин», оказывающий подсушивающее, успокаивающее, противозудное, противовоспалительное действие. После того, как справились с мокнутием, можно применять мази: нафталановую, инталовую, индометациновую.
Очень хорошо зарекомендовало себя такое средство лечения атопического дерматита у детей, как лосьон «Каламин», оказывающий подсушивающее, успокаивающее, противозудное, противовоспалительное действие. После того, как справились с мокнутием, можно применять мази: нафталановую, инталовую, индометациновую.
Не увлекайтесь гормональными мазями! Длительное их применение может вызвать дистрофические изменения кожи. При сильном зуде применяются настои валерианы, листьев мяты, семян укропа, отваров череды, крапивы, подорожника (внутрь).
Для лечения вторичной инфекции используют 1-2% раствор бриллиантового зеленого, 0,5-1% раствор метиленовой синьки, раствор перманганата калия 1:5000, гелиомициновую мазь.
Лучшая мера для профилактики и лечения экссудативного диатеза —длительное грудное вскармливание и создание гипоаллергенной обстановки в квартире.
Задумайтесь, не является ли провоцирующим фактором наличие в доме четвероногого (кошка, собака), летающего (попугайчик, канарейка) или плавающего (рыбки) любимца.
Лимфатико-гипопластический диатез у детей: симптомы и лечение
Такая аномалия конституции, как лимфатико-гипопластический диатез у детей, характеризуется стойким увеличением лимфатических узлов, разрастанием лимфоидной ткани (аденоиды, миндалины), избыточной массой тела и частыми заболеваниями.
Выраженные проявления лимфатико-гипопластический диатеза имеют около 10% детей первого года жизни.
Эти дети обычно бледные, рыхлые, эмоционально вялые, апатичные, отстают в физическом развитии от сверстников, так как мускулатура развита слабо, тонус ее понижен, а кожа дряблая, складчатая. Разрастание лимфоидной ткани в носоглотке приводит к увеличению миндалин и росту аденоидов, что проявляется нарушением носового дыхания, частыми насморками и ухудшением мозгового кровоснабжения. А это в свою очередь ведет к понижению работоспособности, снижению памяти, рассеиванию внимания. Поэтому не удивляйтесь, если воспитательница детского сада говорит, что ваш ребенок на занятиях невнимательный, отвлекается на посторонние дела, плохо усваивает материал. Принимайте меры к оздоровлению, не переносите проблему из детского сада в школу.
Принимайте меры к оздоровлению, не переносите проблему из детского сада в школу.
В выделяемой врачами группе длительно и часто болеющих детей (ДЧБ) основную массу составляют дети с лимфатическим диатезом.
Эти дети наиболее подвержены частым респираторным инфекциям, аденоидиту, гнойному отиту, лимфадениту, хроническому тонзиллиту, ангине, пиелонефриту. Это и понятно, ведь разрастание лимфоидной ткани в носоглотке создает почву для образования очага хронической инфекции. Оттуда микробы проникают по широкой и короткой слуховой трубе непосредственно в уши — вот вам и отит, или с током крови разносятся по организму, вызывая воспаление в отдаленных органах (пиелонефрит).
При обследовании часто выявляется увеличение вилочковой железы, функциональные изменения со стороны сердечно-сосудистой системы, кожные проявления, схожие с высыпаниями при экссудативном диатезе, изменения в клинических и биохимических анализах крови.
Основными моментами в лечении детей с лимфатическим диатезом являются рациональное питание (грудное вскармливание!), соблюдение режима дня, закаливающие процедуры, массаж и гимнастика, санация очагов хронической инфекции.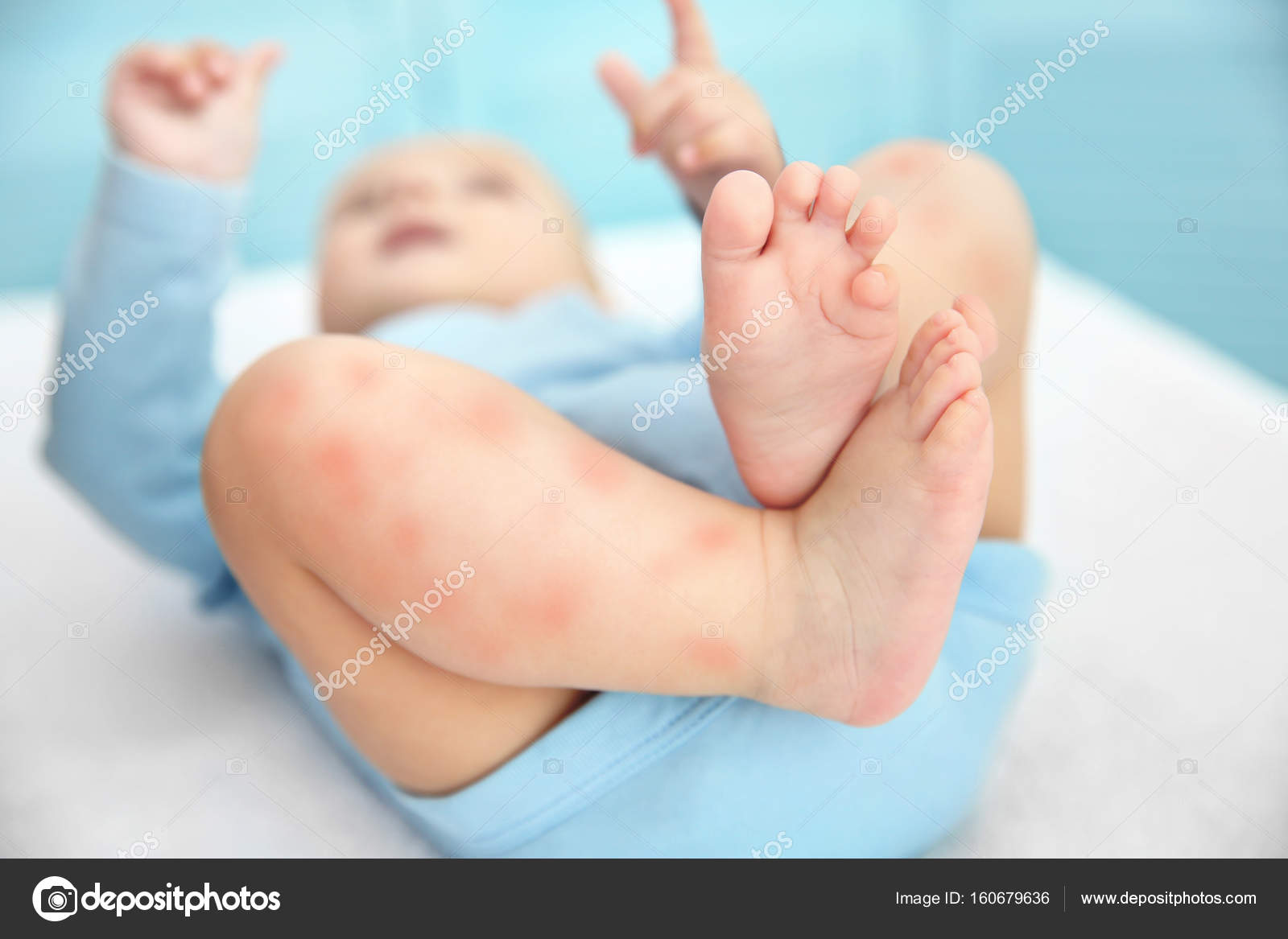
Ребенок должен получать достаточные дозы витаминов, микроэлементов и адаптогенов, стимулирующих защитные силы организма.
Для лечения лимфатико-гипопластического диатеза из современных иммуностимуляторов применяются иммунал, бронхомунал, тимоген, виферон и другие, рекомендованные врачом для конкретного больного.
Нервно-артритический диатез у детей: фото и симптомы
Такая аномалия конституции, как нервно-артритический диатез у детей, имеет своим основным отличительным признаком повышенную нервную возбудимость, отмечаемую у детей практически с рождения и не снижающуюся по мере взросления.
В психическом развитии они намного опережают своих сверстников: рано начинают говорить, самостоятельно учатся читать, имеют хорошую память, легко запоминают длинное стихотворение и способны своими словами пересказать содержание любой книги или телепередачи. Они любознательны и учатся с удовольствием. Окружающие считают их вундеркиндами и прочат им большое будущее.
И лишь родители знают, сколько терпения и выдержки требуется для общения с таким ребенком, имеющим неуравновешенную и возбудимую нервную систему. Он отличается эмоциональной нестабильностью, легким переходом от смеха к слезам и наоборот. Ему не чужды ночные кошмары и боязнь темноты. Сильные внешние раздражители (громкий звук, яркая вспышка света, лай собаки, резкий запах) способны спровоцировать у такого ребенка нервный тик или судорожные сокращения мышц конечностей. Он не переносит многие пищевые продукты, да и вообще любая пища вызывает у него отвращение. Родители водят его по врачам, чтобы хоть как-то наладить аппетит, но анорексия носит стойкий характер и не поддается лечению.
Как видно на фото, дети с нервно-артритическим диатезом имеют низкую массу тела, выглядят стройными и грациозными:
Лишь незначительная часть детей имеет склонность к полноте с ранних лет, а девочки начинают резко набирать вес, вступая в пубертатный период.
Ребенок с нервно-артритическим диатезом капризный, упрямый, стремится настоять на своем и добиться выполнения своих требований любой ценой. Часто жалуется на недомогание и усталость. Наиболее распространенными симптомами нервно-артритического диатеза являются тошнота, боли в животе, в мышцах конечностей, сильные головные боли по типу мигрени, появление кожной сыпи в виде крапивницы, аллергическая реакция на укусы комаров, навязчивый малопродуктивный кашель. Также характерным симптомами нервно-артритического диатеза у детей является ацетонемический криз (ацетонемическая рвота), свидетельствующий о нарушении обменных процессов в организме. Внезапно или после короткого недомогания появляются боли в животе без определенной локализации, тошнота и неукротимая рвота, приводящая к резкой слабости и обезвоживанию. У ребенка изо рта появляется запах ацетона (прелых яблок), говорящий о нарушении кислотнощелочного равновесия в сторону ацидоза.
Наиболее распространенными симптомами нервно-артритического диатеза являются тошнота, боли в животе, в мышцах конечностей, сильные головные боли по типу мигрени, появление кожной сыпи в виде крапивницы, аллергическая реакция на укусы комаров, навязчивый малопродуктивный кашель. Также характерным симптомами нервно-артритического диатеза у детей является ацетонемический криз (ацетонемическая рвота), свидетельствующий о нарушении обменных процессов в организме. Внезапно или после короткого недомогания появляются боли в животе без определенной локализации, тошнота и неукротимая рвота, приводящая к резкой слабости и обезвоживанию. У ребенка изо рта появляется запах ацетона (прелых яблок), говорящий о нарушении кислотнощелочного равновесия в сторону ацидоза.
На этих фото показаны признаки нервно-артритического диатеза у детей раннего возраста:
Развитию криза способствуют острые заболевания, нервные стрессы, насильственное кормление, нарушение режима питания (употребление бобовых, томатов, шоколада, кофе, какао).
Дети с нервно-артритическим диатезом требуют к себе деликатного и бережного отношения, исключения интенсивных психических нагрузок и насильственных воздействий (принуждений, приказаний). В семье необходимо создать доброжелательную атмосферу, исключающую выяснение отношений и разговоров на повышенных тонах. Желательно составить режим дня, чередующий периоды труда и отдыха, умственных и физических занятий, включающий ежедневные прогулки и занятия физкультурой.
Лечение нервно-артритического диатеза у детей, диета и питание
Огромное значение при лечении нервно-артритического диатеза у детей придается диетическому питанию, направленному на предупреждение ацетонемических кризов. В диете ребенка с нервно артритическим диатезом должны преобладать молочные продукты, овощи, фрукты и крупяные изделия. Мясо, птицу и рыбу вводите в рацион 2-3 раза в неделю в виде отварных или тушеных блюд.
Исключите из питания детей с нервно-артритическим диатезом крепкие бульоны, мясо молодых животных (телятина, цыплята), колбасные изделия, грибы, некоторые овощи (шпинат, щавель, спаржу, цветную капусту, бобовые, петрушку), крепкий чай, кофе, какао, шоколад.
При развитии ацетонемического криза, не дожидаясь прихода врача, начинайте борьбу с ацидозом и обезвоживанием, то есть поите ребенка щелочными напитками: регидрон, глюкосолан, оралит, щелочные минеральные воды, 0,5-1% раствор питьевой соды. Выпаивание имеет свои особенности: малыми порциями (чтобы не провоцировать рвоту), но часто. При лечении нервно-артритического диатеза полезны очистительные клизмы, ускоряющие выведение кетоновых тел из организма. С этой же целью показан прием энтеросорбентов (активированный уголь, полифепан, энтеросгель, смекта).
Дети должны периодически получать курсы лечения витаминами (кальция пантотенат, В), препаратами, стимулирующими обменные процессы (оротат калия) и защищающими печень (эссенциале, ЛИВ-52 и др.).
Что такое нервно-артритический диатез у детей
12 декабря 2016 10:00
Такие малыши уже в грудном возрасте более возбудимы, а с возрастом эта особенность усиливается. Карапузы любознательны, оживлены, хорошо запоминают, могут узнать и повторить им показанное, или прочитанное. Частенько детки жалуются на ночные страхи и не хотят спать сами, придумывают себе истории и вымышленных друзей, уписываются но ночам.
Карапузы любознательны, оживлены, хорошо запоминают, могут узнать и повторить им показанное, или прочитанное. Частенько детки жалуются на ночные страхи и не хотят спать сами, придумывают себе истории и вымышленных друзей, уписываются но ночам.
Читайте такжеПочему так важно грудное кормление в первый час жизни
Родители деток жалуются на их чувствительность к запахам, тики, головные боли, навязчивый кашель и беспричинные рвоты. Кожные сыпи не характерны для таких деток, а если и встречаются то в грудном возрасте, на щечках и ягодицах, в виде мелких пузырков, красных, ярких, лакированных участков на коже.
Дети реагируют на режим питания частыми болями в животе. Большая часть таких малышей имеет маленькую массу тела вплоть до анорексии, но некоторые детки склонны к полноте, чаще девочки. Нередко по утрам изо рта запах ацетона, «прелых листьев».
Читайте такжеГрудное молоко защищает детей от болезней
Основной метод лечения — это диета и режим. Карапузов надо беречь от интенсивных психических нагрузок, ограничить просмотр телевизора и компьютера. Полезны систематические закаливания, физическая активность: зарядка, танцы, гимнастика, прогулки, катания на самокате и велосипеде.
Карапузов надо беречь от интенсивных психических нагрузок, ограничить просмотр телевизора и компьютера. Полезны систематические закаливания, физическая активность: зарядка, танцы, гимнастика, прогулки, катания на самокате и велосипеде.
В питании не злоупотреблять мясными и жирными блюдами, шоколадом, бобовыми, какао и кофе.
Не нужно таких малышей кормить насильно! Но больших перерывов в еде следует избегать.
Диета при нервно-артритическом диатезе у детей
- Должны преобладать молочные продукты, овощи и фрукты, гречневая и овсяная крупа, перловка и пшено.
- Ограничивают мясо, птицу, речную рыбу (особенно жаренную, копченную), бульоны, навары, холодцы.
- Полностью исключают мясо молодых животных (телятина, цыплята), субпродукты (мозги, печень,почки), колбасные изделия, грибы, некоторые овощи (щавель, шпинат, спаржа, ревень, цветная капуста, зеленый горошек), все продукты промышленного консервирования на которых нет этикетки «детское питание».
- Под строгим запретом находятся и те продукты, которые способны спровоцировать возбуждение нервной системы — это крепкий чай, соленые и острые блюда, а также пряности, и конечно, кофе. Пользу приносят щелочные минеральные воды, компоты, морсы, цитрусовые соки и вишневый сок.
- На ночь лучше дать ребенку блюда содержащие трудноусвояемые углеводы (гречневая, овсяная каши, овощи, ржаной хлеб, картофель).
Для малышей, полезна фитотерапия — настои трав валерианы, пустырника, пассифлоры.
Безусловно, такие детки справляются с одновременными нагрузками и занятиями по иностранным языкам, музыкой, рисованием, вязанием, хоккеем и так далее, но их не стоит перегружать. Большие нагрузки являются провокатором проявлений не только диатеза но и его возможных последствий (подагры, мочекаменной болезни, ожирения, бронхиальной астмы).
Мочекислый диатез — всегда ли это проявление нарушенного пуринового обмена? | #08/11
Мочекислый диатез часто рассматривается как синоним нервно-артритического диатеза, который является одним из вариантов аномалии конституции. Понятие «конституция» характеризует совокупность морфофункциональных свойств организма ребенка, определяющих индивидуальные особенности его реактивности [1–3]. Диатез, или иначе аномалия конституции, характеризует особенности того или иного вида обмена, которые в определенных условиях могут реализоваться в патологию. Для нервно-артритического диатеза характерна повышенная интенсивность пуринового обмена, конечным продуктом которого является мочевая кислота (МК). Как и другие типы диатезов, мочекислый диатез не рассматривается как патология, а является пограничным состоянием, характеризующимся повышенным риском возникновения ряда заболеваний. При этом виде диатеза имеется склонность к дискинезиям желудочно-кишечного тракта с возникновением ацетонемической рвоты, чаще встречается сердечно-сосудистая патология, заболевания нервной системы, артриты, мочекаменная и желчекаменная болезни, сахарный диабет и др. Сам по себе факт нарушения обмена МК, с точки зрения проф. Н. П. Шабалова, является важным, но не единственным маркером этого вида диатеза, который может предрасполагать к некоторым видам патологии. В этом плане в рамках данной публикации хотелось бы поднять вопрос — всегда ли следует рассматривать мочекислый кристаллурический диатез как мочекислый диатез в связи с нарушением пуринового обмена. Прежде всего, следует определиться, что характеризует мочекислый кристаллурический диатез и отличается ли он от мочекислого диатеза в общепринятом понимании? Основным и определяющим признаком этого вида диатеза являются часто возникающие кристаллы МК или ее солей в осадке мочи. Но этот феномен как раз и является одним из признаков, характеризующих мочекислый диатез, в основе которого лежит избыточное образование МК как проявление напряженности пуринового обмена. Поэтому следовало бы рассматривать мочекислый кристаллурический диатез (МКД) как синоним мочекислого диатеза, а значит, и нервно-артритического диатеза. Причины возникновения кристаллов МК и ее солей различны (рис. 1). В клинической практике нередко встречаются дети, в осадке мочи которых часто обнаруживаются кристаллы МК и ее солей при отсутствии каких-либо признаков нервно-артритического диатеза. Иными словами, отсутствует повышенная нервная возбудимость как в грудном возрасте, так и в последующие возрастные периоды. Отсутствует у них и ускоренное психическое развитие, нет эмоциональной лабильности, нет склонности к ацетозу, а потому у них нет повышенного уровня кетоновых тел, аммиака, МК в крови, т. е. не развивается ацетонемический криз. Обращает на себя внимание и тот факт, что у этих детей отсутствует гиперурикозурия, а мочекислая кристаллурия наблюдается даже при отсутствии повышенной осмоляльности мочи, т. е. кристаллурия возникает в неконцентрированной моче. Это позволяет выделять этот вид диатеза как особую разновидность мочекислого диатеза и, во всяком случае, не рассматривать его как проявление нервно-артритического диатеза. Если нет нарушения пуринового обмена, что же тогда лежит в основе этой разновидности диатеза? В основе развития мочекислого кристаллурического диатеза лежит сниженная способность мочи предотвращать образование кристаллов выделяющейся МК и ее солей. Известно, что моча обладает повышенной растворяющей способностью по сравнению с водой. Это обусловлено тем, что моча представляет собой сложный многокомпонентный раствор, содержащий различные ионизированные элементы кристаллических веществ и крупномолекулярные органические коллоидные субстанции. Взаимодействие между ними обеспечивает повышенную растворимость солей в единице объема [4]. Поэтому даже при высокой концентрации кристаллообразующих солей (в данном случае МК и ее солей — уратов) не происходит образования кристаллов. МК и ее соли, как известно, плохо растворяются в кислой моче и тем хуже, чем ниже рН мочи. При этом их концентрация в моче может быть незначительной, и наоборот, если реакция мочи становится нейтральной или щелочной, выпадения кристаллов не происходит даже в условиях гиперурикозурии. Поэтому мочекислый кристаллурический диатез в отличие от мочекислого диатеза целесообразно рассматривать не как проявление нарушенного пуринового обмена, а как следствие ограниченной способности дистального отдела канальца подщелачивать мочу. Эта способность, как известно, связана с функцией аммониогенеза. Аммониогенез всегда связан с древнейшей функцией канальцевого эпителия — ацидогенезом, при котором происходит синтез ионов водорода (Н+) под действием фермента карбоангидразы, присутствующего в канальцевом эпителии. Образующиеся Н+ секретируются в просвет канальца и тем самым подкисляют мочу. Повышенное образование и секреция их наступает при накапливании в процессе обмена кислых валентностей, что способствует поддержанию кислотно-основного состояния организма. В норме повышенный ацидогенез всегда сопровождается усилением образования аммиака под влиянием фермента глутаминазы, отщепляющей аммиак от глутамина. Аммиак диффундирует в просвет канальца и, соединившись с Н+, образует ион аммония (NH4+), который и подщелачивает мочу. Недостаточная функция аммониогенеза проявляется ацидурией, когда рН мочи стабильно низкий и не превышает 6.0, что способствует выпадению кристаллов МК. Нарушение этой функции может иметь наследственную природу и носить семейный характер. До недавнего времени этот вариант мочекислого диатеза рассматривался как идиопатический. Нарушение функции аммониогенеза может возникнуть вторично на фоне любой почечной патологии с развитием тубулоинтерстициального синдрома.
| Рис. 1. Причины мочекислой кристаллурии |
Таким образом, рассматривая мочекислый диатез с позиций сегодняшнего дня, следует отличать диатез, обусловленный особенностью пуринового обмена, от диатеза, вызванного нарушенной функцией аммониогенеза. При этом важно выделять первичную форму нарушенного аммониогенеза, которая, скорее всего, имеет семейный характер, и именно эта форма и должна характеризовать этот вид мочекислого диатеза, который в отличие от нервно-артритического диатеза целесообразно назвать МКД. Выделение этой разновидности диатеза позволит более рационально осуществлять профилактику возможного перехода данного вида диатеза в мочекислую нефропатию, а далее — в интерстициальный нефрит дизметаболического генеза или мочекаменную болезнь (рис. 2). При этом диатезе нет никакой необходимости, в отличие от нервно-артритического диатеза, строго придерживаться диеты с ограничением продуктов, богатых пуринами, ибо здесь нет нарушения пуринового обмена, а поэтому нет гиперурикемии и гиперурикозурии. Необходимо лишь поддерживать достаточный диурез и способствовать подщелачиванию мочи периодическим назначением цитратов, соответствующих минеральных вод и ряда других препаратов, стремясь поддерживать рН мочи в пределах 6,4–6,8.
Рис. 2. Эволюция мочекислого кристаллурического диатеза |
Мочекислая нефропатия в отличие от МКД — это уже патология, и ее возникновение связывается до сих пор с наличием у таких детей нервно-артритического диатеза, т. е. с нарушением пуринового обмена [5–7]. Однако уратная нефропатия возникает не только в результате нарушения пуринового обмена, когда имеется повышенный эндогенный синтез МК, но и как следствие нарушения транспорта ее в почках, а также из-за низкой ингибиторной способности мочи, реализуемой нарушением образования аммиака дистальным отделом нефрона.
Термин «мочекислая (урикозурическая) или уратная нефропатия» часто используется для определения различных заболеваний, связанных так или иначе с нарушением обмена МК и проявляющихся при этом либо поражением почек, либо только мочевыводящих путей. В первом случае, когда кристаллизация возникает в паренхиме (интерстиции) почек и в ответ на это возникает абактериальный воспалительный процесс, следует говорить об интерстициальном нефрите. Во втором случае, когда кристаллурия возникает лишь в просвете канальцев, это может быть нормой, или характеризовать наличие мочекислого кристаллурического диатеза, или быть проявлением уратной нефропатии.
Основным признаком МКД является возникновение повышенного кристаллообразования. Если кристаллы МК или ее солей образуются в своей массе в просвете канальцев и паренхима почки не поражается, а в осадке мочи имеется только кристаллурия, то это еще не нефропатия. Это может быть нормой, когда при избытке экзогенных пуринов усиливается выделение образующихся мелких кристаллов мочекислых солей и не возникает условий для образования из них микролитов. Когда то же самое наблюдается при отсутствии избытка экзогенных пуринов и нет повышенного эндогенного их образования (нет гиперурикозурии), то следует предполагать наличие МКД.
В практической работе педиатра, а также детского нефролога часто ставится диагноз «мочекислая или уратная нефропатия», если в осадке мочи обнаруживаются кристаллы МК или ее солей. Сам факт наличия кристаллов солей не является основанием для постановки данного диагноза. О дизметаболической нефропатии следует говорить лишь при часто наблюдаемой кристаллурии, сопровождаемой периодически микрогематурией, абактериальной лейкоцитурией, а также дизурией. При этом отсутствует нарушение функции почек, т. к. кристаллообразование и им обусловленный патологический процесс возникают лишь в просвете мочевыделительных путей, не затрагивая воспалительным процессом интерстициальную ткань почек. Однако повышенное образование кристаллов при обменной нефропатии может наблюдаться и в интерстиции почек, что часто обуславливает появление при ультразвуковом исследовании эхоположительных сигналов, которые исчезают при достаточном питьевом режиме. При этом нет воспалительного процесса в интерстиции, возникновение которого будет характеризовать переход обменной нефропатии в интерстициальный нефрит дизметаболического генеза. В этом случае могут быть выявлены нарушения функции почек за счет поражения тубулоинтерстиция в виде снижения концентрационной способности почек, ацидо- и аммониогенеза, что на ранних этапах может быть обнаружено при проведении нагрузочных проб.
Каковы должны быть действия врача, если при обследовании детей с мочекислой кристаллурией не обнаруживается гиперурикемия и гиперурикозурия? При выявлении повышенной кристаллурии необходимо, прежде всего, исключить наличие ацидурии. Для этого следует определить рН в разных по времени собранных порциях мочи. При обнаружении постоянно низких значений рН (< 6,0) для выяснения причины требуется оценить функцию аммониогенеза. Это можно осуществить с помощью проведения нагрузочной пробы с хлористым аммонием (проба Ронга-Девиса) [8]. При этом должен возникнуть компенсированный метаболический ацидоз, в ответ на который усиливается образование и выделение ионов водорода дистальным отделом канальцев и образование аммиака из глутамина путем активации фермента глутаминазы. Поскольку эта проба трудоемка, требует использования сложной и дорогостоящей аппаратуры и плохо переносится больными, была разработана и предложена нагрузочная проба с Лазиксом [8, 9], которая, не вызывая ацидоза, способствует усилению ацидо- и аммониогенеза. Отсутствие или недостаточное образование аммиака в ответ на нагрузку указывает, что причиной ацидурии является сниженный аммониогенез. Эта так называемая идиопатическая форма мочекислой нефропатии фактически является тубулопатией дистального канальца, приводящей к ацидурии.
В связи с этим следует различать две разновидности мочекислого диатеза — диатеза, обусловленного напряженностью пуринового обмена, протекающего с повышенным выделением с мочой МК и ее солей, и диатеза, вызванного сниженной способностью дистального отдела нефрона подщелачивать мочу. Это способствует ацидурии и выпадению кристаллов МК и ее солей при отсутствии гиперурикозурии. Вторую разновидность мочекислого диатеза в отличие от нервно-артритического варианта мочекислого диатеза следует обозначить термином «мочекислый кристаллурический или ацидурический диатез», носящий часто семейный характер и обусловленный сниженной способностью почек подщелачивать мочу, подчеркивая тем самым, что основным признаком его является ацидурия. Таким образом, требуется дифференцированный подход к пациентам, у которых наблюдается уратная кристаллурия. При выявлении ацидурического диатеза нет никакой необходимости ограничивать ребенка, как, впрочем, и взрослого, в приеме продуктов, богатых пуринами. Лечение и профилактика у таких лиц должны быть направлены на повышенный прием жидкости и на подщелачивание мочи. Что касается фитотерапии, то она должна контролироваться не только величиной диуреза, но и обязательно реакцией мочи
Литература
- Маслов М. С. Аномалии конституции (диатезы) в детском возрасте / Многотомное руководство по педиатрии. Под ред. проф. Тур А. Ф. М., 1960, т. 1, с. 471–524.
- Сергеев Ю. С. Конституция человека, конституциональные типы, аномалии конституции и диатезы у детей // Педиатрия. 2005, № 5, с. 67–71.
- Шабалов Н. П. Диатезы и аномалии конституции как педиатрическая проблема // Педиатрия. 2005, № 5, с. 72–76.
- Томах Ю. Ф., Клепиков Ф. А. Кристаллурические диатезы. Харьков, 1992. С. 24–49.
- Юрьева Э. А., Длин В. В. Обменные нефропатии у детей. В кн.: Дисметаболические нефропатии, мочекаменная болезнь и нефрокальциноз у детей. М., 2005. С. 9–72.
- Коровина Н. А., Захарова И. Н., Гаврюшова Л. П. и др. Дисметаболические нефропатии у детей: диагностика и лечение (руководство для врачей). М., 2007. С. 12–16.
- Малкоч А. В. Дисметаболисеские нефропатии и мочекаменная болезнь. В кн.: Практическое руководство по детским болезням. Т. 6: Нефрология детского возраста. Под ред. Таболина В. А., Бельмера С. Б., Османова И. М. М., 2005. С. 472–516.
- Ривкин А. М., Папаян А. В. Лабораторная диагностика: исследование мочи и клиническая оценка результатов. В кн.: Клиническая нефрология детского возраста. Под ред. проф. Папаяна А. В. СПб, 1997. С. 60–80.
- Архипов В. В., Ривкин А. М. Диагностика функционального состояния почек с использованием фуросемида. Методические рекомендации. Под ред. проф. Папаяна А. В. СПб, 1996. 13 с.
А. М. Ривкин, кандидат медицинских наук, доцент
СПбГПМА, Санкт-Петербург
Контактная информация об авторе для переписки: [email protected]
Лицензионные книги по медицине |
| |
Нервно-артритический диатез у детей: симптомы и лечение
Нервно-артритический диатез у детей — опасное заболевание с довольно тяжелым течением и неприятными последствиями.К счастью, заболевание диагностируется не так часто — в разных источниках есть информация о том, что от 2 до 5% детей страдают этим заболеванием. С другой стороны, многие исследователи утверждают, что количество заболевших с каждым годом увеличивается, поэтому родителям следует уделять пристальное внимание состоянию и особенностям развития малыша.
Естественно, многие ищут информацию о том, как, что такое недуг и с какими отклонениями в работе организма он связан. На какие приметы нужно обращать внимание? Существуют ли лекарства, которые могут навсегда избавить от болезни?
Основные характеристики заболевания
Нервно-артритический диатез у ребенка — хроническая патология, которая связана с нарушением нормального обмена веществ.В частности, у маленьких пациентов наблюдается обмен пуриновых нуклеотидов, что приводит к повышению уровня мочевой кислоты. При этом также вовлекаются нарушения обменных процессов жиров и углеводов.
Кроме того, у детей с этим диагнозом наблюдается измененная реактивность нервной системы, что, безусловно, влияет на общее развитие организма, поведение, процесс формирования личности. Этот недуг опасен тем, что увеличивает вероятность развития ожирения, аллергии, калькулезных процессов и других проблем в будущем.Чем раньше он будет диагностирован, тем больше шансов, что он будет успешно исправлен.
Причины и факторы риска
Конечно, первое, что интересует родителей — это причина того или иного заболевания. Нервно-артритический диатез у детей — проблема наследственная. Считается, что данная патология передается генетически от родителей, страдающих нарушением пуринового обмена (подагра, мочекаменная болезнь и др.).
К факторам риска также относятся болезни матери во время беременности, в частности ожирение, сахарный диабет, желчекаменная болезнь.По статистическим данным, у родителей, страдающих нервно-психическими заболеваниями, например неврастенией, шизофренией, шанс родить ребенка с этой формой диатеза выше.
Конечно, есть и другие возможные причины — неправильное питание беременной, употребление слишком большого количества животного белка. Заболевания в младенчестве, кстати, тоже могут быть спровоцированы слишком ранним введением прикорма, особенно мяса и других белковых продуктов. Частичную роль в формировании диатеза играет неблагоприятная экологическая среда.
Первые признаки болезни
На самом деле симптомы этой формы диатеза самые разнообразные, и проявляются они в разные периоды жизни. Их приятно разделить на четыре основные группы — кожный, дисметаболический, спастический и неврастенический синдромы.
На что обращать внимание при подозрении на нейроартритический диатез у детей? Симптомы в младенчестве обычно связаны с нервными расстройствами. Малыш очень возбужден, часто плачет и раздражает, плохо спит, сон очень беспокойный и непродолжительный.
Из-за спазмов гладкой мускулатуры, которые также развиваются на фоне сбоев обмена веществ, у ребенка беспокоят кишечные колики. У таких детей часто бывают затяжные запоры.
Нейроартритический диатез у детей: фото и проявления болезни у детей старшего и подросткового возраста
Остальные признаки (а они очень разные) начинают проявляться по мере взросления ребенка. Для удобства их стоит рассматривать в группах.
Поражения кожи — характерное проявление нейроартритического диатеза у детей. Часто у ребенка развивается экзема и крапивница, атопический дерматит. Кроме того, может появиться ангионевротический отек. Все эти симптомы связаны с аллергическими реакциями.
Что касается спастического синдрома, то он связан с повышенным тонусом мышц, что провоцирует спазм гладкой мускулатуры. Именно поэтому у детей часто развиваются бронхоспазм, запоры, почечная и кишечная колика, гипертония. К симптомам заболевания также относятся стойкие мигрени.Часто дети жалуются на боли в области сердца. Также следует сказать, что у маленьких пациентов иногда развивается бронхиальная астма, которая, к счастью, сравнительно легко поддается лечению.
Кроме того, диатез часто вызывает нарушения обмена веществ. Мочевая кислота постепенно накапливается в суставах и кристаллизуется, вызывая воспаление. Дети часто жалуются на боли в суставах, которые особенно усиливаются ночью.
Одним из самых ярких и опасных проявлений болезни считается ацетонемический криз.В результате употребления большого количества мясных продуктов, а также при интенсивных физических нагрузках, стрессах и эмоциональном перенапряжении в крови и моче появляется ацетон. В дыхании больного ощущается резкий запах ацетона — особенно он сильный по утрам.
Это явление часто сопровождается тошнотой, рвотой и резким снижением аппетита. Кстати, эти симптомы чаще диагностируются у девочек в возрасте от 2 до 10 лет (хотя они могут проявляться и у мальчиков) и в большинстве случаев исчезают в период полового созревания.
Есть также нарушения нервной системы. Из-за повышения уровня мочевой кислоты дети обычно слишком активны, и развиваются намного быстрее, чем их сверстники — это касается как психоэмоционального, так и интеллектуального развития (часто дети с этой формой диатеза намного легче и быстрее учатся в школе. учебный план). Патология сопровождается другими симптомами — это плаксивость, иррациональная раздражительность, резкие смены настроения, повышенная чувствительность к запахам, частые головные боли, проблемы со сном.Часто бывает ночное недержание мочи. К характерным признакам можно отнести значительное увеличение количества каловых масс, которое наблюдается даже при резком снижении аппетита. Дети с диатезом часто страдают анорексией и другими расстройствами пищевого поведения.
Как выглядит процесс диагностики?
К сожалению, диагностировать нейроартритический диатез у ребенка первых месяцев жизни бывает очень редко, так как клиническая картина может быть нечеткой. Расстройства поведения матери и ребенка часто связывают со спазмами желудка и другими детскими проблемами.Тем не менее информативные методы диагностики существуют и применимы даже в младенческом возрасте — это подробный анализ крови и мочи.
При лабораторных исследованиях мочи можно обнаружить наличие различных солей, что указывает на неправильный обмен веществ и возможное образование камня. При исследовании образцов крови можно отметить повышенный уровень мочевой кислоты и креатинина. В дальнейшем необходимы инструментальные исследования, например, УЗИ органов выделительной системы, рентгенограммы и т. Д.
Есть много заболеваний с похожей клинической картиной: холецистит, инфекционный артрит, панкреатит, невроз, ревматизм, пиелонефрит и некоторые другие недуги. Эти патологии часто напоминают нервно-артритический диатез у детей. Диагностика обязательно должна включать тесты и меры, которые могут подтвердить, связаны ли симптомы с диатезом. Только после этого можно составить схему лечения.
Нейроартритический диатез у детей: лечение
К сожалению, это хроническая патология.Это означает, что даже в зрелом возрасте пациент будет по-прежнему подвержен нарушениям обмена веществ, аллергии, мочекаменной болезни и т. Д. Какие меры требует нервно-артритический диатез у детей? Как лечить это расстройство?
Терапия в этом случае длительная и сложная. Здесь важен не только прием лекарств, но и определенный образ жизни, которого нужно придерживаться постоянно. Если речь идет о медикаментозном лечении, то маленьким пациентам в зависимости от симптомов назначают следующие средства:
- Дети часто принимают успокаивающие средства, такие как настойка валерианы, Новопассит, Фтосед и др.
- Препараты, снижающие выработку мочевой кислоты.
- Витаминно-минеральные комплексы, нормализующие обменные процессы (препараты должны содержать калий и кальций).
- Гепатопротекторы (препараты, защищающие клетки печени) — эффективными считаются Эссенциале, Гепабене и Карсил.
- Иногда необходимо принимать нестероидные противовоспалительные препараты, в частности те, которые содержат ибупрофен и парацетамол.
- При поражениях кожи также используются заживляющие мази, а иногда и кремы на основе стероидных гормонов (в тяжелых случаях).
- При возникновении ацетонемического приступа рекомендуется дать ребенку сладкий чай, свежий сок или негазированную щелочную воду.
- Сильные приступы рвоты и развивающееся обезвоживание — показание к приему витамина С и раствора глюкозы.
Нейроартритический диатез у детей требует соблюдения определенного образа жизни. В частности, такому ребенку противопоказаны сильные стрессы, эмоциональные и умственные нагрузки, чрезмерные физические нагрузки. Это, конечно, не означает, что малыша нужно защищать от любых нагрузок.Конечно, ребенок с таким диагнозом может ходить в школу. Плавание, длительные прогулки на свежем воздухе, регулярные массажи и лечебная гимнастика — все это положительно скажется на состоянии организма.
Правильная диета при диатезе
Еще один важный этап коррекции такого заболевания, как нервно-артритический диатез у детей. Диета — залог успеха терапии, и пациенту на протяжении всей жизни следует соблюдать определенные принципы питания. Целью лечебной диеты является сокращение употребления продуктов, содержащих животные белки и пуриновые основания.
Специалисты рекомендуют включать в рацион супы и блюда из овсяной и перловой крупы, гречки и риса. Полезными будут свежие фрукты и овощи, которые необходимо ввести в рацион. Пациенты могут употреблять молочные продукты и свежевыжатые соки.
Нужно ли полностью исключить из рациона животные жиры и белки при таком заболевании, как нервно-артритический диатез у детей? Комаровский, как и другие известные педиатры и исследователи, утверждает, что белки просто необходимы организму растущего ребенка.Поэтому в рацион следует вводить блюда из курицы и нежирной рыбы, но их нужно готовить на пару. В период обострения рекомендую давать щелочную негазированную воду (Боржоми).
Чем опасна болезнь? Возможные осложнения
Многих родителей интересуют вопросы, что такое нервно-артритический диатез у детей. Ключевыми моментами являются симптомы и лечение заболевания, его причины и возможные осложнения. Наличие патологии означает, что дети предрасположены к ряду заболеваний, в частности к ожирению и диабету.Из-за изменения состава мочи повышается риск нефрита, нефропатии и мочекаменной болезни в будущем.
Высокое содержание мочевой кислоты в крови создает условия для развития воспалительных процессов и подагры в зрелом возрасте. Пациенты с таким диагнозом более подвержены атеросклерозу и некоторым другим заболеваниям сердечно-сосудистой системы. Также могут возникать хронические мигрени и неврологические заболевания.
Существуют ли эффективные методы народного лечения?
Конечно, народные лекари предлагают множество средств для борьбы с диатезом.И даже сами врачи находят некоторые рецепты полезными. Например, эффективна яичная скорлупа. Сначала сварить яйцо (варить около 5 минут). Снятую скорлупу необходимо очистить от внутренней пленки и погрузить в кипяток еще на несколько минут. После этого скорлупу сушат (без попадания солнечных лучей) и измельчают в порошок в кофемолке. Рекомендуется взять половину чайной ложки порошка, добавив несколько капель лимонного сока и запив кипяченой водой. Курс лечения длится месяц, после чего нужно сделать перерыв.
Полезным при диатезе считается свежий сок редиса. Сначала ребенку нужно дать каплю сока, постепенно увеличивая объем до одной чайной ложки, а затем и столовой. Также специалисты рекомендуют угощать детей вкусным пюре из кабачков и тыквы, заваривать мятный чай с медом и готовить соки из свежей моркови и малины.
Прогнозы и меры профилактики
Нейроартритический диатез у детей — хроническая патология. Избавиться от него невозможно, но можно предотвратить развитие сопутствующих заболеваний, в частности ожирения, диабета и т. Д.Не стоит отказываться от спокойного размеренного образа жизни даже после достижения совершеннолетия.
Важными средствами профилактики являются посильные, но не слишком интенсивные упражнения, правильное питание, закаливание тела, массажи. У детей и подростков прием лекарств нужно проходить каждые полгода. Конечно, нужно внимательно следить за режимом труда и отдыха, создавать условия для здорового полноценного сна и регулярно проходить медицинские осмотры, помогающие выявить прогрессирование заболевания на ранних его стадиях.
Унаследованные заболевания двигательных нейронов у детей: помимо спинальной мышечной атрофии
Резюме
Заболевания двигательных нейронов у детей включают группу нейродегенеративных заболеваний, характеризующихся началом мышечной слабости и атрофии в возрасте до 18 лет, что связано с потерей двигательных нейронов различными нейронами. сети головного и спинного мозга. Хотя генетические основы разнообразны, успехи в секвенировании следующего поколения изменили диагностические парадигмы.Это укрепило клиническое фенотипирование и молекулярно-генетический опыт, необходимый для управления сложностями таких диагнозов. В свою очередь, улучшенная генетическая технология и последующая идентификация генов позволили глубже понять механизмы дегенерации двигательных нейронов и то, как эти заболевания являются частью спектра нейродегенеративных расстройств. Общие патофизиологии включают нарушения в архитектуре и функции аксонов, процессинг РНК и контроль качества белка. Этот обзор включает в себя обзор клинических проявлений, генетики и патофизиологии унаследованных детских двигательных нейронных заболеваний, помимо классической спинальной мышечной атрофии, связанной с SMN1, и описывает недавние достижения в секвенировании следующего поколения и его клиническое применение.Специфическое модифицирующее заболевание лечение становится клинической реальностью при некоторых нарушениях двигательного нейрона, что подчеркивает важность своевременной и конкретной диагностики.
1. Введение
Заболевания двигательных нейронов (БДН) представляют собой гетерогенную группу прогрессирующих заболеваний, приводящих к затруднениям при ходьбе, движении, дыхании и глотании, с началом заболевания от рождения до взрослого возраста. Признаками являются слабость и атрофия из-за нарушения работы нижнего двигательного нейрона и / или спастичность из-за дисфункции верхнего двигательного нейрона.В глобальном масштабе наиболее распространенная двигательная нейронопатия у детей связана с пикорнавирусными инфекциями, включая вирус Коксаки, энтеровирусы 68 и 71, эховирус и полиомиелит, которые проявляются острым вялым параличом, который может возникать во время вспышек [1, 2]. В то время как у детей с синдромом двигательных нейронов (наследственный, иммуноопосредованный, инфекционный, паранеопластический, спорадический или токсический) могут рассматриваться различные причины, у детей с коварным началом и медленным прогрессированием часто учитывается генетическая основа.Количество выявленных генетических причин, которые демонстрируют вариабельность и совпадение клинических фенотипов, продолжает расти в эпоху геномного тестирования. У детей наиболее распространенным наследственным заболеванием двигательных нейронов является спинальная мышечная атрофия (СМА) из-за гомозиготного нарушения гена выживания двигательного нейрона 1 ( SMN1, ), при этом классические фенотипы требуют целевого тестирования гена SMN1 [3]. Помимо типичного СМА и преобладающих фенотипов нижних мотонейронов, патология может быть более обширной, включая многочисленные гены, связанные с БДН у детей и молодых людей.
В то время как детские болезни двигательных нейронов могут иметь клинические проявления, ограниченные клетками переднего рога, менее распространенные фенотипы SMA плюс или атипичные SMA могут также демонстрировать дополнительные признаки, указывающие на более обширное неврологическое или мультисистемное поражение, включая глухоту, эпилепсию, энцефалопатию, спастичность, нарушение зрения, стволовые, мозжечковые, желудочно-кишечные или ревматологические расстройства [4, 5]. С другой стороны, амиотрофия может развиться позже или действительно может не быть основным проявлением мультисистемных расстройств.Кроме того, может быть очевидным первичное поражение верхнего мотонейрона, называемое ювенильным боковым амиотрофическим склерозом (jALS). Множество причинных генов экспрессируются повсеместно и обеспечивают клиническую, патологическую и генетическую информацию о дегенерации двигательных нейронов у детей как о спектре нейродегенеративных расстройств.
Секвенирование следующего поколения (NGS), включающее панели генов, полное секвенирование экзома (WES) и полное геномное секвенирование (WGS), произвело революцию в парадигме клинического генетического тестирования редких заболеваний.«Нейрогеномика», новая эра генетики в неврологии, критически полагается на тщательное клиническое фенотипирование для точной интерпретации молекулярно-генетических результатов. Здесь мы рассматриваем педиатрические заболевания мотонейронов, выходящие за рамки СМА, и представляем их отличительные клинические и невропатологические особенности вместе с анализом причинных генов и механизмов заболевания, лежащих в основе дегенерации моторных нейронов. При возможности оценивается конкретное модифицирующее заболевание лечение, что подчеркивает важность конкретного генетического диагноза.Мы включили синдромы «СМА плюс», при которых дисфункция двигательных нейронов является основным, но не единственным признаком, ювенильный боковой амиотрофический склероз (jALS) и мультисистемные расстройства, при которых очевидна амиотрофия, связанная с дегенерацией нижних мотонейронов. Наследственные спастические параплегии и чистые дистальные наследственные моторные нейропатии были подробно рассмотрены и поэтому не описаны [6–8]. Кроме того, мы также предоставляем обзор технологий NGS, которые могут использоваться в молекулярно-генетических диагностических подходах.В настоящее время они трансформируют клиническую практику и могут предоставить метод для своевременной и рентабельной диагностики, имеющей решающее значение для проведения терапии на как можно более ранней стадии для этих разрушительных групп расстройств. Подчеркивается важность мультидисциплинарного подхода к преодолению сложностей молекулярно-генетической диагностики.
2. Клинические проявления, молекулярная генетика и терапия, изменяющая конкретное заболевание
Клинический подход к педиатрическому пациенту с синдромом двигательных нейронов включает оценку паттерна слабости, амиотрофии и прогрессирования (проксимального или дистального, бульбарного или респираторного). вовлечение), наличие спастичности и глубоких сухожильных рефлексов, а также семейный анамнез.Обычные электрофизиологические исследования и электромиография (ЭМГ) важны для установления нейрогенной основы и дифференциации от миопатии и нарушений нервно-мышечного соединения. При заболеваниях нижних мотонейронов, сложные мышечные потенциалы действия (CMAP) снижаются при нормальной или слегка замедленной скорости моторной проводимости, что соответствует потере аксонов. ЭМГ может демонстрировать активную денервацию с положительными резкими волнами и потенциалами фибрилляции и / или многофазными многофазными двигательными единицами высокой амплитуды длительной продолжительности, что указывает на реиннервацию.Для получения подробного фенотипа важны оценки зрения, слуха, познания / развития и общих систем. Нейровизуализация головного мозга и позвоночника, а также офтальмологические и аудиологические исследования могут дополнять клиническую оценку. Тестирование второго уровня может включать биопсию мышц или нервов, метаболические тесты или тестирование одного гена на основе фенотипа. Технологии NGS трансформируют клиническую практику и все чаще используются в качестве диагностического теста первого уровня для своевременной диагностики ().Это усиливает важность точной клинической оценки вместе со знанием различных клинических и генетических фенотипов, чтобы иметь возможность поставить молекулярно-генетический диагноз.
Диагностический подход к пациенту (<18 лет) с заболеванием двигательных нейронов (БДН). БАС: боковой амиотрофический склероз; СК: креатининкиназа; dHMN: дистальная наследственная моторная нейропатия; GBS: синдром Гийена-Барре; БДН: расстройство двигательного нейрона; МРТ: магнитно-резонансная томография; NGS: секвенирование нового поколения. ∗ геномное тестирование панелью; WES или WGS могут быть вариантом в зависимости от доступности в вашем регионе.
3. Синдромы СМА плюс
Синдромы СМА плюс, или атипичная СМА, охватывают расстройства, при которых дисфункция нижних мотонейронов является первичным, но не единственным признаком и может быть классифицирована по различным паттернам мышечной слабости (суммировано в). СМА с понтоцеребеллярной гипоплазией (SMAPCh2 / PCh2), СМА с прогрессирующей миоклонической эпилепсией (SMAPME), врожденная СМА с артрогрипозом и переломами и СМА, вызванная митохондриальными нарушениями, демонстрируют преобладающую проксимальную слабость.СМА с преобладанием нижних конечностей (SMALED) и лопаточно-малоберцовая СМА имеют отчетливые паттерны слабости, как описано. Синдром Брауна-Виалетто-Ван Лаэра (BVVL) и связанные с ним расстройства имеют выраженную бульбарную слабость.
Таблица 1
Спинальная мышечная атрофия плюс синдромы с известными вариантами генов.
| Спинальная мышечная атрофия плюс синдром | Возраст начала | Клинический фенотип | Ген | Функция гена | Тип наследования | Ссылки |
|---|---|---|---|---|---|---|
| Понтоцеребеллярная гипоплазия со спинальной мышечной атрофией (ПЧ2) | Ранний младенец | Тяжелая гипотония, арефлексия, мышечная слабость, центральное нарушение зрения, дисфагия, дыхательная недостаточность и приобретенная микроцефалия.МРТ головного мозга показывает гипоплазию мозжечка с различным поражением моста. | VRK1 EXOSC3 EXOSC8 TSEN54 SLC254A6 | Процессинг РНК | AR | [13, 15, 17–19, 149] |
| MORC2 | Ремонт митохондриальной ДНК, процессинг РНК, метаболизм липидов | |||||
| Спинальная мышечная атрофия с прогрессирующей миоклонической эпилепсией (SMAPME) | Детство (начальное развитие нормальное) | Слабость проксимальных мышц, гипотония, арефлексия и мышечное истощение.Фасцикуляция языка и может иметь нейросенсорную тугоухость, полиартикулярный артрит и слабость лица. Поздняя миоклоническая эпилепсия | АСАх2 | Архитектура цитоскелета — разветвление аксонов Аутофагия (лизосомы) | AR | [24, 25] |
| СМА с аномалиями скелета | ||||||
| СМА с врожденным артрогрипозом и переломами | Антенатальный | Артрогрипоз, переломы, пороки сердца, тяжелая гипотония, слабость, арефлексия с фасцикуляцией языка и дыхательная недостаточность.Ранняя смерть | SMN1 TRIP4 ASCC1 УБА1 | Процессинг РНК | AR | [33, 34] |
| Спинальная мышечная атрофия, Х-сцепленная (SMAX2) | Антенатально | Врожденные переломы, артрогрипоз и фасцикуляция языка | УБЭ1 | Расщепление протеина через протеасомы | Х-сцепленное | |
| Летальный артрогрипоз с поражением клеток переднего рога (LAAHD) | Антенатально | Последовательность деформации акинезии плода.Смерть в утробе матери или в течение нескольких дней после родов. Нормальный спинной мозг. Финский | GLE1 | Процессинг РНК — медиатор экспорта мРНК | AR | [40] |
| Синдром летальной врожденной контрактуры 1 (LCCS1) | Антенатальный | Летальный исход во внутриутробном периоде, наиболее тяжелая форма артрогрипоза. Аномальный спинной мозг с выраженным истончением. Финский. | GLE1 | Процессинг РНК — медиатор экспорта мРНК | AR | [40] |
| Синдром летальной врожденной контрактуры 2 (LCCS2) | Антенатальный | Врожденные контрактуры, дисморфизм и поражение мочевого пузыря. Ранняя смерть большинства. Израильские бедуины | ERBB3 | Модулятор процессинга РНК фосфатидилинозитол-3-киназы / пути Akt | AR | [44] |
| Расстройства, связанные со СМА плюс митохондрии | ||||||
| Кардиоэнцефаломиопатия с дефицитом цитохром С оксидазы (CEMCOX1) | Младенчество | Фенотип СМА с гипертрофической кардиомиопатией, судорогами, задержкой психомоторного развития и офтальмоплегией.МРТ головного мозга — аномалии белого вещества и базальных ганглиев | SCO2 | Структура и функция митохондрий | AR | [45] |
| Синдром митохондриального истощения 2 (MTDP2) | Младенчество / детство | Гипотоническая мышечная слабость, дыхательная недостаточность, задержка психомоторного развития с судорогами и офтальмологией. Прогрессивная и широкая вариативность | ТК2 | Функция митохондрий — истощение митохондриальной ДНК | AR | [48] |
| Синдром митохондриального истощения 3 (MTDP3) | Младенчество | SMA с младенческим началом, дисфункцией печени, | Функция митохондрий — истощение митохондриальной ДНК | AR | [47] | |
| SMA плюс с характерными слабыми сторонами | ||||||
| Спинальная мышечная атрофия с преобладанием нижних конечностей 1 (SMALED1) | Врожденная для взрослых | Непрогрессивная, проксимальная> только дистальная слабость ноги МРТ нижней конечности — без приводящих и полусухожильных мышц бедра | DYNC1h2 | Цитоскелетная динамика — динеиновый комплекс для аксонального транспорта | AD | [53] |
| Спинальная мышечная атрофия с преобладанием 2 нижних конечностей (SMALED2) | Врожденная до взрослого | Медленное прогрессирование дистальных слабых ног,> руки> с некоторыми контрактами | BICD2 | Динамика цитоскелета динеин-динактиновый комплекс | AD | [54, 55] |
| Лопаточно-перистая спинальная мышечная атрофия (SPSMA) | Ранний взрослый | Прогрессирующая слабость лицевых и пекторальных мышц гортани.Возможна нейросенсорная глухота, аномалии скелета. МРТ нижней конечности — без двуглавой мышцы бедра и медиальной икроножной мышцы | TRPV4 | Кальциевый канал | AD | [62] |
| Врожденная дистальная мышечная атрофия позвоночника (DSMA) | Врожденная | Непрогрессирующая слабость и контрактуры только проксимальных и дистальных отделов ног | TRPV4 | Кальциевый канал | AD | [62] |
| Спинальная мышечная атрофия с респираторным дистресс-синдромом (SMARD) | Младенчество | Дистальный> проксимальный отдел нижней конечности> слабость верхних конечностей с ранней диафрагмальной слабостью и дыхательной недостаточностью | Рибосомный биогенез в процессинге РНК | AR | [72, 73] | |
| Спинальная мышечная атрофия с респираторным дистрессом 2 (SMARD2) | ЛАСИЛИЯ | Х-сцепленный | [64] | |||
| Синдром Брауна-Виалетто-Ван Лаэре (BVVL) | От раннего детства до взрослого возраста | Прогрессирующий понтобульбарный паралич со слабостью рук, кистей и лица; атаксия; дисфагия; истощение языка; и фасцикуляции и нейросенсорная глухота | SLC52A3 SLC52A2 UBQLN1 | Транспорт витаминов Расщепление белков через протеасомы | AR | [74, 79, 80] |
3.1. Понтоцеребеллярная гипоплазия со спинальной мышечной атрофией (PCh2)
Понтоцеребеллярная гипоплазия (PCH) относится к группе тяжелых нейродегенеративных заболеваний, характеризующихся спиноцеребеллярной дегенерацией, пороками развития червя и передней доли мозжечка и тяжелой гипоплазией мозга. На основании клинических и патологических проявлений выделено десять подтипов ПКГ, при этом тип 1 ПКГ ассоциирован с клеточной болезнью передних рогов, напоминающей детскую спинальную мышечную атрофию [9–12].Мутации, разделяющиеся по аутосомно-рецессивному принципу в генах киназы 1, связанной с осповакциной ( VRK1 ), компонента экзосомы 3 ( EXOCS3 ) и компонента экзосомы 8 ( EXOCS8 ), являются причиной генов PCH типа 1A, типа 1B и типа 1С соответственно. Большинство пациентов с PCh2 (от 30 до 75 процентов) имеют патогенные варианты в EXOCS3 , с мутациями в VRK1 и EXOCS8 , ограниченными только несколькими описаниями случаев [12-15]. Генетическая этиология еще предстоит выяснить у дополнительных пациентов с PCh2, и NGS может предоставить возможности для дальнейшего понимания патогенеза.
TSEN54 Мутации составляют> 90% описанных аллелей, связанных с PCh3 и PCh5, однако также были описаны в единственном случае с PCh2 [16]. Последний экспрессируется в стволе мозга и мозжечке, но не в клетках переднего рога, и ожидается подтверждение в других семьях. В 2016 году с помощью WES были идентифицированы еще два гена SMAPCH. К ним относятся мутации в митохондриальном белке профсекции SLC254A6 , о которых сообщалось в двух семьях с летальной понтоцеребеллярной гипоплазией с атрофией зрительного нерва и сенсомоторной нейропатией [17], а также мутация de novo MORC2 у одного пациента [18].
Клиническими характеристиками, связанными с «классическим» PCh2, являются тяжелая гипотония, арефлексия, мышечная слабость, центральное нарушение зрения, дисфагия, дыхательная недостаточность и приобретенная микроцефалия с клиническим началом в первые месяцы жизни и смерть в раннем младенчестве. Атаксия, нистагм и задержка психомоторного развития могут быть дополнительными признаками. Антенатальное начало с врожденными контрактурами и многоводием, при тяжелых фенотипах и более позднее начало при более мягких фенотипах, с выживанием после взросления, также описано [11, 15].МРТ головного мозга показывает гипоплазию мозжечка с поражением полушарий мозжечка вместе с различным поражением моста и головного мозга. Генерализованная атрофия коры головного мозга может наблюдаться у пациентов, выживших дольше. Электрофизиология демонстрирует сенсомоторную аксональную невропатию и острую денервацию на электромиографии (ЭМГ).
Имеются корреляции генотип-фенотип в пределах патогенных вариантов EXOCS3 , связанных с клиническим исходом, выживаемостью и степенью гипоплазии моста.Пациенты с гомозиготными миссенс-мутациями p.D132A имеют более мягкий фенотип, лучшую сохранность моста и выживаемость [14]. Пациенты с гомозиготными мутациями p.G135E или измененными мутациями рамки считывания демонстрируют тяжелые фенотипы и смерть в детстве.
ПЧ2А и ПЧ2Б демонстрируют отчетливые клинические проявления с вариабельностью. Тяжелая врожденная микроцефалия, ранняя СМА, легкая или умеренная умственная отсталость и выживаемость до позднего детства являются характеристиками PCh2A [15].Фенотип PCh2A расширился и включил периферическую невропатию, микроцефалию с упрощенным круговым паттерном, спастичность и нормальную когнитивную функцию [19, 20]. Напротив, фенотип PCh2B обычно включает тяжелую умственную отсталость с минимальным овладением речью или без нее, приобретенную и прогрессирующую микроцефалию, глазодвигательную дисфункцию и отсутствие фиксации [12]. Редкое раннее начало осложненной спастической параплегии с легкой понтоцеребеллярной гипоплазией было признано вариантом синдрома PCh2B [21], что дает клиническое представление о более обширной дисфункции двигательной сети.Фенотип PCh2C включает тяжелую мышечную слабость и неспособность нормально развиваться с задержкой психомоторного развития, часто с нарушением зрения и слуха и респираторными проблемами, вызывающими преждевременную смерть. МРТ головного мозга показывает признаки гипоплазии мозжечка, гипоплазии мозолистого тела и гипомиелинизации [13].
VRK1 кодирует серин / треонин киназу, связанную с регуляцией клеточного цикла, модификацией гистонов, реакциями репарации ДНК и нарушением процессинга РНК, которые являются общими патофизиологическими темами, лежащими в основе заболеваний двигательных нейронов [22].Точно так же нарушение метаболизма мРНК из-за дисфункции экзосом лежит в основе PCh2B и C, при этом EXOSC3 и EXOSC8 кодируют основные компоненты экзосом РНК, которые обрабатывают и разрушают РНК, регулируя активность экспрессии генов. Однако, в то время как мутации EXOSC3 затрагивают в основном двигательные нейроны спинного мозга и клетки Пуркинье, клетки олигодендроглии также являются мишенями мутаций в EXOSC8 [13].
На сегодняшний день не разработана модифицирующая болезнь терапия SMAPCh2, и лечение симптоматическое, включая нутритивную и респираторную поддержку, как указано.
3.2. Спинальная мышечная атрофия с прогрессирующей миоклонической эпилепсией (SMAPME)
SMAPME впервые была описана Jankovic и Rivera в 1979 году [23]. Он характеризуется начинающейся в детстве прогрессирующей слабостью проксимальных мышц, гипотонией, арефлексией и истощением мышц после начальных этапов нормального развития [24]. Могут присутствовать легкая слабость лица, фасцикуляции языка, нейросенсорная тугоухость и тремор. Рестриктивные респираторные заболевания и рецидивирующие инфекции возникают с прогрессирующей слабостью и дисфагией.Миоклоническая эпилепсия, резистентная к лечению, возникает позже по ходу болезни, а выживаемость ограничивается первыми двумя десятилетиями из-за дыхательной недостаточности. Редкие фенотипические варианты, описанные недавно, включают полиартикулярный артрит и спинальную мышечную атрофию, легкий фенотип СМА без миоклонической эпилепсии, эпилептический миоклонический статус век и более поздние проявления в подростковом возрасте с абсанс-эпилепсией и атоническими припадками [25–28]. МРТ головного мозга в норме. ЭМГ демонстрирует активную денервацию с положительными острыми волнами и потенциалами фибрилляции вместе с большой амплитудой, большой продолжительностью и многофазными двигательными единицами, соответствующими реиннервации ().Электроэнцефалограф (ЭЭГ) может быть нормальным или может демонстрировать медленный фон с эпилептической активностью, которая может быть светочувствительной. Эпилептическая активность клинически может соответствовать подергиванию конечностей, киванию головой или отрицательному миоклонусу [24, 29].
Клинические картины синдрома СМА плюс. (а) ЭМГ, демонстрирующая многоамплитудную многофазную моторную единицу, характерную для реиннервации. (b) Увеличение H&E × 200, показывающее маленькие круглые денервированные мышечные волокна ∗ и большие гипертрофированные мышечные волокна ∗∗ .(c) Атрофия дистального отдела ноги. (d) Рентгенография, показывающая эвентрацию диафрагмы при спинальной мышечной атрофии с респираторным дистрессом (SMARD. (e) Атрофия языка при синдроме Брауна-Виалетто-Ван Лаэра. (f) Истощение рук с контрактурами, наблюдаемое при синдроме Брауна-Виалетто-Ван Лаэра.
Мутации в N-ацилсфингозинамидогидролазе 1 ( ASAh2 ), кодирующей лизосомный фермент кислую церамидазу, были идентифицированы WES как основа аутосомно-рецессивной формы SMAPME [24]. Интересно, что мутации в ASAh2 также связаны с мутациями ASAh2 . Липогранулематоз Фарбера, тяжелое раннее начало лизосомного нарушения накопления, поражающее несколько тканей, характеризующееся накоплением сфинголипидов.Уровни кислой церамидазы in vitro составляют <10% при болезни Фарбера по сравнению с <30% при SMAPME, что коррелирует с фенотипической изменчивостью. Чжоу и др. [24] продемонстрировали функциональное влияние миссенс-мутаций ASAh2 in vivo, при котором снижение активности кислой церамидазы вызывает заметные дефекты моторно-аксонального ветвления и значительное увеличение апоптоза в спинном мозге. Кроме того, церамиды являются предшественниками сложных сфинголипидов, которые важны для нормального функционирования как развивающегося, так и зрелого мозга и миелина.
В настоящее время для SMAPME не существует модифицирующего болезнь лечения. Миоклоническую эпилепсию можно лечить противоэпилептическими средствами, но обычно она устойчива к традиционной терапии. Трансплантация кроветворного костного мозга использовалась при других лизосомных заболеваниях и болезни Фарбера [30]. Как и многие другие лизосомные расстройства накопления с тяжелым заболеванием ЦНС, трансплантация гемопоэтических стволовых клеток не принесла облегчения неврологических проявлений. Трансплантация аллогенных стволовых клеток успешно почти полностью разрешила гранулемы у детей с болезнью Фарбера, с преобладающим системным заболеванием и без неврологических нарушений, что привело к улучшению подвижности и контрактур суставов [31, 32].
3.3. СМА с врожденным артрогрипозом и переломами
Клинические признаки инфантильной СМА с артрогрипозом отражают антенатальную слабость, сопровождающуюся нарушениями в развитии костей и сердца. Это включает в себя уменьшение шевеления плода, многоводие, тазовое предлежание, гипоплазию легких с диафрагмальной эвентрацией и раннюю смерть. Артрогрипоз, остеопения, множественные переломы и врожденные пороки сердца обычно присутствуют при рождении.Присутствуют выраженная гипотония, мышечная слабость, арефлексия и фасцикуляции языка. Проблемы с дыханием, кифоз, сколиоз, легкая микрогнатия и снижение выражения лица являются результатом мышечной слабости. Электрофизиология и мышечная патология подтверждают денервацию (). Большинство детей не выживают более нескольких месяцев из-за дыхательной недостаточности без обширной респираторной и медицинской помощи. На сегодняшний день не известна модифицирующая болезнь терапия врожденной СМА с артрогрипозом или переломами.
Рецессивные мутации в четырех различных генах были идентифицированы при врожденной / младенческой СМА с артрогрипозом или переломами, включая двигательный нейрон выживания 1 ( SMN1 ) [33], рецептор рецептора тироидного гормона 4 ( TRIP4 ), коинтегратор сигнала активации 1 субъединица 1 комплекса ( ASCC1 ) [34] и убиквитин-подобный модификатор-активирующий фермент 1 ( UBA1 ). В совокупности эти расстройства исключительно редки, с аутосомно-рецессивным или Х-сцепленным рецессивным ( UBA1 ) наследованием, а описания приведены в отчетах о случаях [34–39].
3.4. Смертельный артрогрипоз с заболеванием клеток переднего рога (LAAHD)
LAAHD — аутосомно-рецессивное заболевание, первоначально описанное в 11 финских семьях, связанное с рецессивными патогенными вариантами посредника экспорта РНК, GLE1 [40, 41]. Клинические признаки включают неподвижность плода с множественными дистальными, внутренними спиральными контрактурами суставов, низко посаженные уши, гипоплазию челюсти, короткую шею, некоторые гипоплазии легких, умеренную водянку и гибель плода во время беременности или в раннем постнатальном периоде [41].Размер и форма спинного мозга нормальные, но двигательные нейроны передних рогов кажутся дегенерированными и уменьшенными в количестве. Головной мозг и мозжечок не поражены. GLE1 представляет собой эволюционно консервативный ген, кодирующий белок, необходимый для экспорта мРНК из ядра в цитоплазму, таким образом регулируя экспрессию гена на нескольких этапах, включая экспорт ядерной мРНК, инициацию трансляции и прекращение трансляции.
В дополнение к LAAHD, варианты GLE1 распознаются при БАС с аутосомно-доминантным наследованием и аутосомно-рецессивным синдромом летальной врожденной контрактуры 1 (LCCS1).LCCS1 имеет наиболее тяжелый фенотип с крайней гипоплазией скелетных мышц и истончением спинного мозга из-за малого количества клеток переднего рога. Это заболевание до сих пор наблюдалось только в Финляндии и обычно приводит к летальному исходу до 32 недель беременности, проявляясь полной неподвижностью, тяжелой водянкой и задержкой внутриутробного развития [41, 42].
Синдром летальной врожденной контрактуры 2 (LCCS2) также включает краниальные и глазные аномалии и резкое увеличение мочевого пузыря, сопровождающееся гидронефрозом и кистозными изменениями почек.Большинство заболевших были мертворожденными или умерли вскоре после рождения из-за дыхательной недостаточности, а вскрытие подтвердило патологию клеток переднего рога. Фенотипическая вариабельность этого синдрома была обнаружена у двух сестер-подростков в возрасте 12 и 13 лет с артрогрипозом, амиотрофией, парезом лица, миопией и дегенеративной витреоретинопатией [43]. Патогенные варианты гена гомолога 3 вируса эритробластного лейкоза v-erb-b2 ( ERBB3 ) были описаны в этом родственном израильско-бедуинском семействе с сегрегацией LCCS2 как аутосомно-рецессивным признаком [44].
3.5. Синдромы СМА плюс, вызванные митохондриальными нарушениями
СМА, вызванная митохондриальными нарушениями, обычно проявляется тяжелой инфантильной СМА, или заболеванием Верднига-Хоффмана, характеризующимся гипотонией, слабостью и последующей дыхательной недостаточностью. Клинические проявления более обширны и могут включать гипертрофическую кардиомиопатию в младенческом возрасте, печеночную недостаточность, спастичность, синдром Ли с энцефалопатией, дисфункцию ствола мозга, судороги и задержку психомоторного развития, птоз и офтальмоплегию [45].Биохимические особенности могут включать лактоацидоз и повышение креатининкиназы. Биопсия мышц может показать групповую атрофию, сниженное окрашивание цитохром-С-оксидазой (ЦОГ) и низкую активность ферментного комплекса дыхательной цепи.
Гены, которые, как известно, вызывают фенотипы СМА при митохондриальных нарушениях, это SCO2 , TK2 и DGUOK . SCO2 кодирует один из белков сборки ЦОГ, связанных со вставкой меди в холофермент, участвующий в биогенезе субъединицы ЦОГ II, который важен для функции внутренней митохондриальной мембраны, в то время как последние два представляют собой синдромы митохондриального истощения, характеризующиеся снижением количество митохондриальной ДНК, нарушающее синтез комплексов дыхательной цепи.Дисфункция митохондрий связана со многими нейродегенеративными состояниями, включая заболевания двигательных нейронов, при которых потребности двигательных нейронов в энергии удовлетворяются за счет окислительного фосфорилирования митохондрий. Наблюдаются отчетливые корреляции генотип-фенотип, и возраст начала и прогрессирования может быть модулирован патогенностью конкретной мутации, особенно при заболеваниях, вызванных составными гетерозиготными вариантами. SCO2 Мутации связаны с детской кардиоэнцефаломиопатией с поражением белого вещества головного мозга и базальных ганглиев, нейрогенной мышечной атрофией и гипертрофической кардиомиопатией [45, 46]; DGUOK также известен как гепатоцеребральный синдром (MTDPS3 — синдром истощения митохондриальной ДНК 3) и характеризуется СМА с дисфункцией печени в младенческом возрасте, нистагмом, церебральной атрофией и смертью от печеночной недостаточности в возрасте до 12 месяцев [47].Интересно, что у взрослых заболевание нижних мотонейронов, прогрессирующая внешняя офтальмоплегия, птоз и миопатия пояса конечностей также описаны с мутациями DGUOK . TK2 связан с синдромом истощения митохондриальной ДНК 2 (MTDPS2), обычно связанным с миопатией с широкой клинической вариабельностью, которая включает внутрисемейную вариабельность, когда в паре братьев и сестер у брата были электрофизиологические данные, совместимые со SMA, и он был жив в возрасте 4 лет, в то время как его сестра умерла в 2 года от сильной слабости и гипотонии с первых месяцев жизни [48].Специфическая терапия СМА, вызванная митохондриальными нарушениями, в настоящее время представлена общей терапией митохондриальных нарушений и направлена на изменение митохондриальной динамики за счет усиления функции дыхательной цепи и устранения токсичных метаболитов. Клиническая реакция на эти методы лечения наблюдалась на животных моделях, но польза для людей еще не доказана. Добавка меди для мутаций SCO2 , которые, хотя и не изменяют мозговые и мышечные симптомы, показала обратимость гипертрофической кардиомиопатии [49].
3.6. Спинальная мышечная атрофия с преобладанием нижних конечностей (SMALED)
SMALED обычно характеризуется врожденным или ранним началом, часто в возрасте до 5 лет, проксимальной> дистальной преобладающей мышечной слабостью нижних конечностей и атрофией с нормальной чувствительностью, часто приводящей к значительное нарушение подвижности. Клинические признаки включают замедленную ходьбу, походку вразвалку, затруднения при ходьбе и потерю дистальных рефлексов. Могут быть очевидны врожденный артрогрипоз, фасцикуляции, деформации стопы с пяточно-вальгусной, плоской и вальгусной ногами (), контрактуры, дисплазия тазобедренного сустава или гиперлордоз [50].Расстройство статическое или умеренно прогрессирующее, и степень истощения мышц не позволяет предсказать степень слабости. После первоначального клинического и патологического описания болезни клеток переднего рога фенотип расширился, включив спастичность и когнитивные нарушения, что указывает на дисфункцию нейронов более широкой двигательной сети у некоторых людей [51]. Кроме того, распознаются проявления во взрослом возрасте или вовлечение верхних конечностей с крылатой лопатки и минимальной дистальной слабостью кисти [51, 52].
МРТ верхних и нижних конечностей демонстрирует характерный паттерн замещения мышечного жира и избирательную гипертрофию. В нижней конечности было диффузное поражение прямой мышцы бедра, латеральной широкой мышцы бедра, промежуточной широкой мышцы бедра, передней большеберцовой мышцы и икроножной мышцы с относительной сохранностью медиальных аддукторов, полусухожильной и малоберцовой мышц. Корковые мальформации лобной и перисильвиальной областей были описаны у пациентов с нарушением интеллекта SMALED1 при нейровизуализации [51].На сегодняшний день не было разработано модифицирующей болезнь терапии SMALED.
методами NGS установлены аутосомно-доминантные мутации гена тяжелой цепи 1 цитоплазмы 1 динеина ( DYNC1h2 ) [53] или гена моторного адаптера бикаудального D-гомолога 2 ( BICD2 ) [54, 55] как причины SMALED 1 и SMALED2, соответственно. Эти белки важны для транспорта аксонов, и выявленные функциональные нарушения показывают их роль в заболевании двигательных нейронов. DYNC1h2 кодирует белок тяжелой цепи динеина, который формирует решающее ядро цитоплазматического динеинового комплекса, который необходим для ретроградного транспорта аксонов, поддержания гомеостаза белка и миграции нейронов.Сходным образом белок BICD2 взаимодействует с моторным комплексом динеин-динактин, также важным для транспорта аксонов. Описаны мутации как моторного, так и хвостового домена в DYNC1h2 [53, 56]. Патогенные варианты в DYNC1h2 также могут быть аллельными для других неврологических расстройств, включая болезнь Шарко-Мари-Тута; аксональный тип 20, CMT20 и умственная отсталость; и аутосомно-доминантный 13 с дефектами миграции нейронов, MRD13. У некоторых пациентов с MRD13 наблюдаются клинические признаки периферической невропатии [57].Совсем недавно синдром варианта BICD2 включает врожденный множественный артрогрипоз с двусторонней перисильвиальной полимикрогирией и фенотип с хронической миопатией [58, 59].
3,7. Спинальная мышечная атрофия лопатки (SPSMA)
SPSMA характеризуется нейрогенной лопаточно-перонеальной амиотрофией, врожденным отсутствием мускулов, прогрессирующей атрофией лопатки и надкостницы и прогрессирующей дистальной слабостью и амиотрофией. Дополнительные отличительные признаки могут включать паралич гортани и случайную нейросенсорную глухоту, низкий рост, сколиоз и легкую дисплазию скелета конечностей и позвонков.Недавно был описан фенотип, сочетающий скапулоперонеальную спинальную мышечную атрофию и скелетную дисплазию, а также межсемейную фенотипическую изменчивость, при которой у отца была обнаружена легкая форма SPSMA, в то время как у сына была обнаружена при рождении скелетная дисплазия и прогрессирующее течение, что, возможно, предполагает прогрессирование заболевания в поколениях [60, 61].
SPSMA представляет собой аутосомно-доминантное заболевание, вызванное мутациями в гене временного рецепторного потенциала ваниллоидного 4 ( TRPV4 ) гена канала. TRPV4 кодирует проницаемый для кальция неселективный катионный канал, экспрессируемый во многих тканях и типах клеток, включая клетки гладких мышц и кости. В периферической нервной системе этот ген экспрессируется в кожных сенсорных рецепторах, в ганглиях задних корешков и, в меньшей степени, в мотонейронах. Таким образом, мутации в TRPV4 являются аллельными для ряда различных заболеваний, включая врожденную спинальную мышечную атрофию и артрогрипоз, аутосомно-доминантную аксональную болезнь Шарко-Мари-Тута типа 2C, дистальную наследственную моторную нейропатию [62, 63] и ряд скелетных заболеваний. дисплазии [64].Клинические и МРТ признаки TRPV4 — спинальная мышечная атрофия, непрогрессирующая, преобладающая нижняя конечность, незначительно отличаются от DYNC1h2 — и BICD2 -SMALED; например, деформациями являются эквиноварусная мышца, а не пяточно-вальгусная, а МРТ нижних конечностей показывает сохранение двуглавой мышцы бедра и медиальной икроножной мышцы, а не приводящих мышц бедра и полусухожильной мышцы [65].
3.8. Спинальная мышечная атрофия с респираторным дистрессом (SMARD)
SMARD — редкое аутосомно-рецессивное заболевание двигательных нейронов, характеризующееся преимущественно слабостью дистальных мышц с параличом диафрагмы, приводящим к тяжелой дыхательной недостаточности, приводящей к преждевременной смерти.Симптомы обычно проявляются на ранней стадии от шести недель до шести месяцев. У пораженных младенцев задержка внутриутробного развития, слабый плач, сосание или врожденная деформация стопы являются первыми симптомами, за которыми следует дыхательная недостаточность из-за паралича диафрагмы () и прогрессирующая мышечная слабость. Однако сообщалось о высокой вариабельности проявлений в семье, о позднем начале и медленно прогрессирующей форме [66, 67]. Описана дальнейшая периферическая невропатия без поражения дыхательных путей [68, 69].Другие клинические признаки, указывающие на патологию, выходящую за рамки СМА, включают сенсорную и вегетативную дисфункцию (чрезмерное потоотделение, задержку мочи, запоры и сердечную аритмию), судороги и прогрессирование пареза черепных нервов [70]. Гистологические и ультраструктурные данные икроножного нерва показали дегенерацию Валлера и выраженную атрофию аксонов с гипогипермиелинизацией и вариабельностью толщины миелина [71]. Эти изменения также распространяются от спинного мозга на двигательные нервы.
SMARD1 вызывается гомозиготными или сложными гетерозиготными мутациями в гене, кодирующем иммуноглобулин μ -связывающий белок 2 ( IGHMBP2 ) [72].Ген экспрессируется повсеместно и кодирует АТФазу / геликазу, отвечающую за разделение двухцепочечной ДНК и РНК. IGHMBP2 Ген состоит из 15 экзонов, и мутации распределены по всем 15 экзонам гена, но чаще встречаются в экзонах 10 и 12. Однако точная роль белка и корреляция между типом мутации и фенотипом остаются неясными. [70]. Различия в клинических фенотипах и исходы могут быть связаны с уровнями белка IGHMBP2.Единственный случай со сходным фенотипом, вызванный X-связанной рецессивной мутацией в белке биогенеза рибосом LAS1L , обозначен как SMARD2 [73].
3.9. Синдром Брауна-Виалетто-Ван Лаэра (BVVL) и другие бульбарные синдромы СМА
Синдром Брауна-Виалетто-Ван Лаэра — редкое аутосомно-рецессивное неврологическое заболевание понтобульбарных мотонейронов (VII, IX, X, XI и XII) с прогрессирующей лицевой слабость, дисфагия, амиотрофия языка (), фасцикуляции и нейросенсорная глухота в детском возрасте [74, 75].Потеря слуха обычно предшествует возникновению неврологического синдрома, который также включает атрофию зрительного нерва, слабость рук (), называемую «ребенок в бочке», и атаксию. Течение болезни прогрессирующее, но непостоянное. Дыхательная недостаточность может наступить в течение 6–18 месяцев в случаях с ранним началом, приводя к рецидивирующим инфекциям грудной клетки и смерти. Сообщается о выживаемости до 6-го десятилетия жизни, а совсем недавно был признан быстрый понтобульбарный паралич у взрослых [76, 77].
Электрофизиология демонстрирует отсутствие слуховых ответов ствола мозга и сенсомоторную нейроопатию [78].У некоторых пациентов наблюдается повышение уровня ацилкарнитинов со средней длиной цепи в сыворотке и дефицит флавина. BVVL вызывается гомозиготными или сложными гетерозиготными мутациями в SLC52A2 , SLC52A3 или гетерозиготной формой в генах UBQLN1 [74, 79, 80]. Гены SLC52A2 и SLC52A3 кодируют транспортеры рибофлавина RFVT2 и RFVT3, и переменная экспрессия обнаруживается в тонком кишечнике, головном мозге плода и спинном мозге. Постулируется, что в основе фенотипа BVVL лежит дефицит нейронального рибофлавина, при этом рибофлавин играет решающую роль в метаболизме углеводов, аминокислот и липидов.Другими общими чертами мутаций SLC52A2 являются сенсомоторная нейропатия и атрофия зрительного нерва [79].
Важно отметить, что генетическая основа BVVL обеспечила целевую терапевтическую стратегию с пероральным или внутривенным введением высоких доз рибофлавина с обычной дозой 10 мг / кг / день. Клинический ответ может быть различным, от быстрого улучшения двигательной функции; сила, слух, зрение, отлучение от груди или прекращение респираторной поддержки; нормализация нейрофизиологических параметров в течение нескольких дней с постепенным улучшением в течение 12 месяцев, стабилизация клинического состояния или, в редких случаях, отсутствие ответа [75, 81].Все аномальные профили ацилкарнитина и уровни флавина нормализовались после приема рибофлавина.
Другие унаследованные бульбарные моторные синдромы включают синдром Фацио-Лондэ, который похож на BVVL, но без потери слуха. Этиология вторична по отношению к патогенным вариантам SLC52A3 . Отдельно синдром Уорстера-Засухи представляет собой врожденный непрогрессирующий супрабульбарный парез, связанный с Х-сцепленным или аутосомным наследованием. Локусы генов еще не идентифицированы. Перисильвийская полимикрогирия может быть идентифицирована при нейровизуализации.В то время как бульбоспинальная мышечная атрофия (BSMA или болезнь Кеннеди) обычно возникает у взрослых, иногда в подростковом возрасте она может проявляться мышечными судорогами, затруднениями жевания и аномальным половым развитием [82]. Более поздние признаки болезни Кеннеди включают бульбарную слабость, слабость проксимальных конечностей, широко распространенные и заметные фасцикуляции, потерю сенсорной функции в дистальных отделах конечностей и нечувствительность к андрогенам. BSMA — это Х-сцепленное рецессивное нейродегенеративное заболевание, вызванное увеличением количества повторов CAG в рецепторе андрогенов.Основное клеточное нарушение, вызываемое мутантным рецептором андрогена AR, связано с нарушением регуляции транскрипции, со вторичными изменениями, происходящими в передаче клеточных сигналов, митохондриальной активности и аксональном транспорте.
4. Детские мультисистемные расстройства, включая болезнь двигательных нейронов. Они обобщены, некоторые из них описаны ниже и выбраны из-за большей заболеваемости, но остаются редкими заболеваниями.Тщательная клиническая, электрофизиологическая и патологическая оценка выявляет прогрессирующую слабость, амиотрофию и денервацию, даже если они не могут быть основными признаками, служащими для точной характеристики фенотипов и руководства лечением.
Таблица 2
Педиатрические мультисистемные расстройства с поражением двигательных нейронов.
| Спинальная мышечная атрофия плюс синдром | Возраст начала | Клинический фенотип | Ген | Функция гена | Тип наследования | Ссылки |
|---|---|---|---|---|---|---|
| Начальное косоглазие, гипотония, слабость и задержка в развитии.Последующий психомоторный регресс, спастическая тетраплегия, дистония, нистагм, атрофия зрительного нерва | PLA2G6 | Структура и функция митохондрий | AR | [90] | ||
| Синдром Олгроува (AAAS) | От детства до взрослого возраста | Надпочечниковая недостаточность, алакрима, ахалазия. Может развиться БАС-подобное расстройство, периферическая сенсомоторная аксональная нейропатия, вегетативная дисфункция, спастичность, спиноцеребеллярный синдром, судороги, умственная отсталость или деменция +/- ладонно-подошвенный гиперкератоз, микроцефалия | AAAS | Регуляторный белок | AR | [98] |
| Синдром Чедиака-Хигаши (CHS) | От раннего до взрослого | Полинейропатия, умственная отсталость, гипопигментация, светобоязнь и системные заболевания (диологические нарушения иммунитета) | LYST | Аутофагия — лизосомальный регулятор трафика | AR | [100] |
| Дистальный SMA с энцефалопатией | Ранний | Дистальная моторная нейропатия, спастическая атаксия и ювенильное начало накопления железа в мозге | Полимеризация микротрубочек на аппарате Гольджи | AR | [91] | |
| Алопеция, прогрессирующие неврологические дефекты и эндокринопатия (ANE) | 2-я декада | Алопеция, тяжелая центральная умственная отсталость, прогрессирующая двигательная активность надпочечниковая недостаточность, низкий рост, микроцефалия и гиподонтия.На МРТ гипоплазия гипофиза | РБМ28 | Процессинг РНК — регуляция биогенеза рибосом | AR | [150] |
| Спиноцеребеллярная атаксия 3 типа (болезнь Мачадо-Джозефа) | Со 2-й декады | Атаксия, лицевые и язычные фасцикулы и различные пирамидальные расстройства , дистония, паркинсонизм, нарушения сна, ригидность, периферическая невропатия, атрофия дистальных мышц или наружная офтальмоплегия | ATXN3 (большие расширения) | Контроль качества протеина — убиквитиновая протеасомная система | AD | [151] |
| Лейкоэнцефалопатия с дистонией и моторной нейропатией | 2–4-я декада | Дистония, лейкоэнцефалопатия, нейропатия задней части мозга, лейкоэнцефалопатия , и азооспермия | SCP2 | Аутофагия — пероксисомальный фермент | AR | [152] |
| Двигательная нейронопатия с катарактой и аномалиями скелета | От раннего до взрослого | Слабость в дистальных отделах руки и проксимальных отделах ног, катаракта, короткое основание черепа, низкорослый кости запястья | АЛДх28А1 | Биосинтез аминокислот | AD | |
| Синдром Драве с моторной невропатией | Младенчество | Эпилептическая энцефалопатия, атаксия, задержка развития, вальгусная стопа.Позднее возникновение аномалий походки — может наблюдаться приседание, двигательная нейропатия | SCN1A | Функция ионного канала | AR | [153] |
4.1. Детская нейроаксональная дистрофия (INAD)
Классическая детская нейроаксональная дистрофия (INAD) — это тяжелое нейродегенеративное заболевание, обычно начинающееся в возрасте до 3 лет. Первоначально он характеризуется косоглазием, гипотонией и задержкой развития, поэтому варианты СМА могут рассматриваться среди дифференциальных диагнозов, поддерживаемых слабостью и денервацией.Впоследствии развиваются психомоторная регрессия, спастическая тетраплегия и планарные реакции разгибателей, дистония, гипорефлексия нижних конечностей и вегетативная дисфункция. Офтальмологические находки также включают нистагм, несогласованные движения глаз и атрофию зрительного нерва со снижением остроты зрения [83–85]. В конечном итоге мышечная слабость вызывает затруднения с кормлением и дыханием, что приводит к аспирации и пневмонии, так что большинство пациентов умирают в возрасте до 10 лет [86]. Припадки не распространены, но при их наличии на интерктальной ЭЭГ может наблюдаться быстрая активность и всплески диффузных спайк-волновых комплексов ().Типичная ЭЭГ во время припадка может состоять из диффузных, нерегулярно резких и высоковольтных медленных волновых комплексов с последующей десинхронизацией [85, 87]. Патологическим признаком являются сфероиды аксонов, тубуловезикулярные структуры и митохондриальные аномалии центральных и периферических нервов [88]. Они могут быть обнаружены внутри миелинизированных и немиелинизированных внутримышечных нервов или периферических нервных окончаний кожи и конъюнктивы. Дегенерация затрагивает также кортикоспинальный тракт. Заметная атрофия мозжечка наблюдалась почти у всех пациентов с PLAG26 при нейровизуализации [83, 89] () с коварной атрофией нижнего червя мозжечка, наличием Т2-взвешенного гиперсигнала коры мозжечка и гипоинтенсивностью бледного шара и субстанции. черная (обнаруживается на более поздних стадиях и объясняется отложением железа).МРТ-спектроскопия также дает некоторые важные ключи к диагностике, когда исследования выявили восстановление N-ацетиласпартата и присутствие лактата в базальных ганглиях [85].
Клинические картины мультисистемных расстройств у детей с поражением двигательных нейронов. (а) Интерриктальная ЭЭГ, показывающая диффузные спайковые и медленные волновые комплексы при детской нейроаксональной дистрофии (INAD). (b) МРТ головного мозга в сагиттальной плоскости, показывающая атрофию мозжечка, наблюдаемую при INAD. (c) Бариевый глоток демонстрирует расширение пищевода и его сужение в соответствии с ахалазией при синдроме AAA.
Гомозиготные мутации в PLA2G6 связаны со спектром нейродегенеративных фенотипов: INAD, атипичная нейроаксональная дистрофия и PLA2G6 -связанные дистония и паркинсонизм. Ген PLA2G6 картируется на длинном плече хромосомы 22q12.3-13.2, кодируя кальций-независимый фермент фосфолипазу A2. Он локализуется в митохондриях, и этот фермент имеет решающее значение для поддержания гомеостаза клеточной мембраны, передачи сигналов, передачи сигналов кальция, пролиферации клеток и апоптоза [90].Новый ген у детей с дистальной моторной нейропатией, спастической атаксией и ювенильным началом накопления железа в мозге был недавно идентифицирован и вызван двуаллельными мутациями в TBCE , важными для полимеризации микротрубочек в аппарате Гольджи, и его нарушение приводит к фрагментации Гольджи. ранее наблюдали в дегенерирующих мотонейронах при БАС [91].
4.2. Синдром AAA
Achalasia-Addisonianism-Alacrima (AAA, тройной А или синдром Олгроува) был впервые описан в 1978 году.Алакрима является самой ранней и наиболее устойчивой особенностью, и другие офтальмологические находки могут включать атрофию или бледность зрительного нерва, высокий астигматизм и анизокорию [92]. Дополнительные мультисистемные особенности включают ахалазию, гастропарез, глоссит, надпочечниковую недостаточность, гиперпигментацию, ладонно-подошвенный гиперкератоз, низкий рост, остеопороз и микроцефалию. Неврологические проявления могут быть разнообразными и прогрессирующими и могут включать в себя БАС-подобное расстройство, периферическую сенсомоторную аксональную невропатию, вегетативную дисфункцию, спастичность, спиноцеребеллярный синдром, судороги (возможно, связанные с гипогликемией), умственную отсталость или слабоумие с началом в детстве или во взрослом возрасте [ 93–95].Связанные с нарушением двигательных нейронов бульбарные проявления могут включать дизартрию, дисфагию, спастичность языка и носовую речь. Повышенный тонус, гиперрефлексия и ненормальное время проведения центральной моторной проводимости отражают дисфункцию кортикальных мотонейронов, хотя обычная МРТ является нормальной [95].
Рецессивные мутации в гене AAAS , кодирующем белок аладин или адракалин, связаны с синдромом AAA. В основном это бессмысленные мутации, приводящие к экспрессии усеченного белка.Отсутствует корреляция генотип-фенотип в AAAS с межсемейной вариабельностью в возрасте начала заболевания. Ген AAAS экспрессируется повсеместно; однако особенно обильная экспрессия наблюдается в надпочечниках, структурах желудочно-кишечного тракта, гипофизе, мозжечке и легких плода [96]. Функция Aladin не совсем понятна, но кодируемый белок принадлежит к семейству регуляторных белков WD-repeat и является частью комплекса ядерных пор, экспрессируемых как в нейроэндокринных, так и в церебральных структурах.Нуклеопорины участвуют в транспорте между ядром и цитоплазмой, таким образом, играя важную роль в клеточном цикле и экспрессии генов периферической и центральной нервной системы [97, 98].
4.3. Синдром Чедиака-Хигаши (CHS)
Синдром Чедиака-Хигаши был впервые описан в 1943 году и представляет собой редкое аутосомно-рецессивное системное заболевание, поражающее гематологическую и неврологическую системы, кожу и глаза. Первый характеризуется гепатоспленомегалией, лимфаденопатией, панцитопенией и тяжелым иммунодефицитом с рецидивирующими гнойными инфекциями и склонностью к кровотечениям.Ускоренная фаза проявляется гемофагоцитарным лимфогистиоцитозом. Частичный глазокожный альбинизм может возникать из-за дефектов пигментации. Неврологическое поражение варьирует, описана двигательная нейронопатия, сопровождающаяся восходящими подошвенными ногами [99]. Черепная и периферическая нейропатия, когнитивные нарушения, судороги, атаксия, спиноцеребеллярная дегенерация и экстрапирамидные признаки, такие как паркинсонизм и деменция, также могут возникать и обычно проявляются поздно [100]. Патогномоничным признаком CHS являются гигантские лизосомные пузырьки внутри всех типов клеток.Результаты МРТ головного мозга неспецифичны, может наблюдаться церебральная или мозжечковая атрофия, которая коррелирует с более длительной продолжительностью заболевания и более тяжелыми когнитивными нарушениями. Недавно наблюдались морфологические изменения задней черепной ямки, где угол, охватывающий верхний червь мозжечка, был более острым у пациентов с ХСН [101]. Хотя трансплантация костного мозга может облегчить гематологические и иммунологические осложнения и продлить выживаемость после 7 лет, неврологический фенотип не изменяется [102].Леводопа может помочь при паркинсонизме. CHS вызывается гомозиготными мутациями в гене регулятора лизосомного трекинга, CHS1 / LYST [103]. Он экспрессируется повсеместно и, как известно, участвует в контроле экзоцитоза секреторных лизосом.
5. Ювенильный боковой амиотрофический склероз (jALS)
Боковой амиотрофический склероз (БАС) состоит из прогрессирующей, хотя и переменной, дегенерации кортикоспинального тракта, ствола мозга и спинномозговых мотонейронов с распространенностью 0.4–2,6 на 100 000 населения [104]. В настоящее время это считается мультисистемным расстройством с различными клиническими проявлениями и преобладающими двигательными симптомами. Клиническое течение также разнообразно, и естественное течение заболевания зависит от возраста и пола. Приблизительно 90% случаев БАС в настоящее время считаются спорадическими, связанными со сложными полигенными факторами и факторами окружающей среды, со средним возрастом начала заболевания в шестом десятилетии, соотношением мужчин и женщин 1,7: 1 и средней выживаемостью с момента появления симптомов в двух случаях. –4 года [105]. Вместе с вариабельной пенетрантностью генетическое консультирование затруднено.Напротив, наследственный БАС в равной степени поражает молодых людей, мужчин и женщин, и имеет бимодальную продолжительность болезни. Примечательно, что ювенильный БАС с возрастом до 25 лет чаще является семейным, чем взрослые формы, и описаны как аутосомно-доминантные, так и рецессивные формы (-ALS 2, 4, 5, 6, 6–21 и 16). . Традиционно семейный БАС с ювенильным началом ассоциировался с более медленным прогрессированием и увеличением выживаемости, чтобы обеспечить нормальную продолжительность жизни [106, 107]. Совсем недавно были выявлены отдельные и явно спорадические фенотипы с агрессивными проявлениями и течением в подростковом возрасте, связанными со специфическими мутациями, например, слитым в саркоме геном ( FUS или ALS6) или супероксиддисмутазой 1 ( SOD1 ). [108].Интересно, что патология ювенильного БАС может также распространяться за пределы двигательной системы с очевидной эмоциональной лабильностью и несоответствующим плачем или смехом (псевдобульбарные признаки), подобно спектру расстройств БАС-ЛТД у взрослых, что подтверждает представление о мультисистемных нейродегенеративных расстройствах и распространенных явлениях. патомеханизмы.
Таблица 3
Ювенильный боковой амиотрофический склероз.
| Спинальная мышечная атрофия плюс синдром | Возраст начала | Клинический фенотип | Ген | Функция гена | Тип наследования | Ссылки | Детство | Спастичность и слабость мышц лица и конечностей с бульбарными или псевдобульбарными симптомами.Возможны когнитивные нарушения. Медленно прогрессирующий. | Альсин | Передача сигналов GEF | AR | [111] |
|---|---|---|---|---|---|---|
| Боковой амиотрофический склероз 4 (ALS4) | Молодой человек | Тяжелая дистальная слабость и пирамидные признаки. Сохранение бульбарной функции и познания. Медленное прогрессирование. | Сенатаксин | Восстановление ДНК и процессинг РНК | AD | [114] |
| Боковой амиотрофический склероз 5 (ALS5) | 1–2 декада | Прогрессирующая дистальная слабость и амиотрофия.Позже спастичность и бульбарные симптомы. Инвалидная коляска связана в 20–40 лет. | Спатаксин | Поддержание аксонов | AR | [116] |
| Боковой амиотрофический склероз 6 (ALS6) | В среднем 40 лет, но может быть несовершеннолетним | Быстро прогрессирующая спастическая походка, слабость, амиотрофия, дизартрия, дизартрия + / — лобно-височная деменция или когнитивные нарушения | FUS | Транскрипция, процессинг РНК и репарация ДНК | AR или AD | [120] |
| Боковой амиотрофический склероз 6-21 (ALS6-21) | Детство | Отдельная семья с выраженной спастичностью, дистальные верхние и нижние конечности слабость и бульбарная дисфункция.Птоз, слабость лица и гинекомастия | Неизвестно , 6p25 и 21q22 | Неопределенно | AR | |
| Боковой амиотрофический склероз 16 (ALS16) | Младенчество или детство | Единственная семья в Саудовской Аравии, затем поражение верхних и нижних конечностей. | SIGMAR1 | Стресс эндоплазматического ретикулума и дисфункция митохондрий | AR | [128] |
5.1. Боковой амиотрофический склероз 2 (БАС 2)
БАС 2 характеризуется началом в детстве, в среднем 6,5 лет, и прогрессирующей спастичностью мышц лица и конечностей с бульбарными или псевдобульбарными симптомами. Последнее может включать неконтролируемый смех и / или плач и спастическую дизартрию. Также могут быть очевидны умеренная атрофия мышц и когнитивные нарушения [107]. БАС-2 очень медленно прогрессирует, и продолжительность жизни может быть нормальной, только некоторые люди теряют способность передвигаться в возрасте от 12 до 50 лет. Выявлены вариабельные фенотипы, связанные с более молодым возрастом начала, генерализованной дистонией и быстрым прогрессированием, спастическим квадрипарезом, микроцефалией и мозжечковыми симптомами [109, 110].
Рецессивные патогенные мутации гена Alsin , хромосома 2q33, вызывают БАС2. Альсин кодирует два белковых транскрипта путем альтернативного сплайсинга, длинную форму, которая включает три предполагаемых домена фактора обмена гуанина (GEF), и короткую форму. Большинство мутаций вызывают усечение длинной и / или короткой формы белка алсина с последующей потерей функции. Альсин вырабатывается во многих тканях, но больше всего в головном мозге, особенно в мотонейронах. Удаленная мутация в длинной форме вызывает ювенильный первичный боковой склероз, тогда как мутация, которая затрагивает как длинную, так и короткую форму, вызывает ALS2 [111].GEFs активируют членов суперсемейства Ras GTPases, чтобы они играли роль в процессах переноса пузырьков и мембран. Несмотря на то, что появились разнообразные функции алсина, очень мало известно о его роли в верхних двигательных нейронах. Фенотип ALS2 также включает восходящую наследственную спастическую параплегию с младенческим началом и ювенильный первичный боковой склероз [111, 112].
5.2. Болезнь моторных нейронов с ювенильным началом с доминантным наследованием и отсутствием бульбарного поражения (ALS4)
ALS4 характеризуется ювенильным началом, часто в возрасте до 25 лет (средний возраст начала 17 лет), с медленным прогрессированием, тяжелой дистальной мышечной слабостью, пирамидной признаки и сохранение познания, бульбарной и сенсорной функции.Начальными симптомами обычно являются затруднения походки из-за поражения нижних конечностей, и ожидаемая продолжительность жизни может быть нормальной [106, 113].
Аутосомно-доминантное наследование с мутациями, картированными в хромосоме 9p34 в гене сенатаксина ( SETX ), вызывает ALS4. Белок сенатаксин обладает ДНК- и РНК-геликазной активностью и играет роль в клеточной регуляции ДНК и РНК, нарушение которой приводит к нейродегенерации [114]. Мутации SETX являются аллельными с атаксией и глазодвигательной апраксией типа 2, а также были описаны при спиноцеребеллярной атаксии 1 с аутосомно-рецессивным наследованием [115].
5.3. Ювенильный боковой амиотрофический склероз 5 (ALS5)
ALS5, очень похожий на ALS1, составляет 40% аутосомно-рецессивных jALS и характеризуется медленно прогрессирующей нижней двигательной нейронопатией, связанной со спастичностью. Начало — между первым и вторым десятилетием (в среднем 16,3 года) с начальной дистальной слабостью и атрофией кистей и стоп, за которыми в конечном итоге следует бульбарная дисфункция [116, 117]. Прогрессирующее заболевание верхних мотонейронов со временем становится более очевидным, и большинство пациентов становятся прикованными к инвалидной коляске с продолжительностью заболевания 20-40 лет (в среднем 34.3 года). У этих пациентов не отмечается никаких сенсорных или когнитивных изменений, а также истончения мозолистого тела.
ALS5 наследуется аутосомно-рецессивно и был сцеплен с областью 6 сМ хромосомы 15q15.1 – q21.1 как с составными гетерозиготными, так и с гомозиготными мутациями, описанными в гене spatacsin [116]. Этот ген повсеместно экспрессируется в нервной системе, преимущественно в мозжечке, коре головного мозга, гиппокампе и шишковидной железе. Белок спатаксин играет роль в поддержании аксонов нейронов, включая транспортировку внутриклеточного груза.Этот ген был впервые описан при спастической параплегии 11 с истончением мозолистого тела (SPG11) и L-допа-чувствительным паркинсонизмом и недавно обнаружен при болезни Шарко-Мари-Тута, аксональной и типе 2X [118, 119].
5.4. Боковой амиотрофический склероз 6 с лобно-височной деменцией или без нее (ALS6)
Агрессивные проявления JALS с ранним началом и быстрое течение заболевания с прогрессирующей спастической походкой, дизартрией, поражением лицевых мышц и дисфункцией нижних мотонейронов связаны с мутациями -в гене саркомы ( FUS или ALS6).Поскольку до 5% семейного БАС связано с мутациями гена FUS , доминантным или рецессивным наследованием и идентификацией у лиц со спорадическим БАС [108, 120] и БАС с паркинсонизмом или лобно-височной деменцией, FUS стал важным генетическим фактором. фактор в ландшафте БАС. Были идентифицированы устойчивые корреляции генотип-фенотип FUS ; напр., мутации p.S96del связаны с большей выживаемостью, в отличие от тяжелого клинического течения со смертью или постоянной вентиляцией легких 15.Через 4 месяца после появления мутации p.P525L [108, 121, 122]. По отдельности, начальная симметричная слабость проксимальных мышц конечностей и сгибателей / разгибателей шеи и минимальное вовлечение верхних мотонейронов связаны с мутацией R521C [123]. Хотя у большинства пациентов когнитивные способности нормальные, могут присутствовать умственная отсталость и трудности в обучении [121, 122, 124].
Патологическим признаком БДН, ассоциированной с FUS , являются базофильные включения в двигательных нейронах [125, 126]. FUS является повсеместно экспрессируемым, преимущественно ядерным, ДНК / РНК-связывающим белком, который образует цитоплазматические агрегаты и импортируется в ядро с помощью транспортатина; он участвует в метаболизме ДНК и РНК и считается протеинопатией.
5.5. Ювенильный боковой амиотрофический склероз 6-21 (ALS6-21)
Сообщалось о единственной кровной семье с четырьмя из шести затронутых детей, обозначенной как ALS6-21, в возрасте от 4 до 10 лет с выраженными чертами верхних мотонейронов, отмеченными истощение дистальных мышц верхних и нижних конечностей и бульбарная дисфункция. Кроме того, были отмечены асимметричный птоз, умеренная слабость лица, умеренная потеря чувствительности и гинекомастия с медленным прогрессированием и потерей способности передвигаться через десять лет.Обнаружено аутосомно-рецессивное наследование с очагами на хромосоме 6p25 и 21q22 [127].
5.6. Ювенильный боковой амиотрофический склероз 16 (JALS16)
JALS16 относится к описанию единственной кровной семьи в Саудовской Аравии, в которую входили шесть больных с медленно прогрессирующей спастичностью и слабостью нижних конечностей и дебютом в возрасте от 1 до 2 лет. Поражение верхних конечностей и потеря способности передвигаться произошли к 20 годам с нормальными респираторными, бульбарными, когнитивными, сенсорными и сфинктерными функциями [128].
Рецессивные мутации в гене рецептора сигма-1, SIGMAR1 , вызывают JALS16. Это шаперон эндоплазматического ретикулума, связывающийся с широким спектром лигандов, а также функционирующий в регуляции ионных каналов, таких как калиевый канал, и может модулировать высвобождение нейротрансмиттеров и транспорт кальция в митохондриях. Считается, что он вызывает патологию двигательных нейронов за счет изменения морфологии эндоплазматического ретикулума, дестабилизации липидного рафта и дефектных эндолизосомных путей [129].Это также необходимо для правильного митохондриального транспорта аксонов в мотонейронах; в частности, ретроградное движение митохондрий и мутантный белок SIGMAR1 снижает продукцию митохондриального АТФ, ингибирует активность протеасом и вызывает повреждение митохондрий, усугубляя вызванную стрессом гибель нейронов эндоплазматического ретикулума [130]. Хотя SIGMAR1 экспрессируется повсеместно, он обогащен мотонейронами ствола и спинного мозга с субклеточным постсинаптическим увеличением [131].Рецессивные мутации SIGMAR1 могут также вызывать дистальную SMA2 и лобно-височную долевую дегенерацию [132, 133]. Кроме того, полиморфизм гена SIGMAR1 также вовлечен в болезнь Альцгеймера, шизофрению, инсульт, амнезию и депрессию [134].
5.7. Другие генетические причины ювенильного БАС
Как и следовало ожидать, количество генов, связанных с jALS, продолжает увеличиваться, а фенотипы среди известных генов ALS и других нейродегенеративных заболеваний расширяются [20, 25].Мутации в Ubiquilin 2 были идентифицированы при Х-сцепленном доминантном БАС с началом у подростков и взрослых и БАСБП [135]. Патология характеризуется убиквитинированными включениями, которые могут колокализоваться с белками TDP-42 и FUS. Кроме того, диагностический обзор в случаях с медленным прогрессированием и длительной выживаемостью может помочь в фокусировке генетического тестирования. Например, повторная нейровизуализация у пациента с jALS идентифицировала отложение железа в базальных ганглиях, направляя секвенирование генов, связанных с нейродегенерацией с накоплением в головном мозге (NBIA) и подтверждая сложную гетерозиготную мутацию гена C19ORF12 , который кодирует трансмембранный митохондриальный белок [136].
5.8. Лечение jALS
Ювенильный БАС остается без лечения; однако многопрофильные специализированные клиники повысили выживаемость и качество жизни взрослых пациентов с акцентом на поддерживающую терапию [137, 138]. Возможная терапевтическая значимость гипервозбудимости коры головного мозга, патофизиологической движущей силы БАС у взрослых и частично улучшающейся рилузолом (единственная терапия, модифицирующая заболевание при БАС), не оценивалась у ювенильных фенотипов БАС [139, 140]. Достижения в понимании патофизиологии БАС продолжают открывать новые терапевтические мишени, при этом генная терапия с использованием антисмысловых олигонуклеотидов демонстрирует эффективность на доклинических моделях и входит в фазу 1 клинических испытаний конкретных генетических подтипов БАС [141, 142].Соответственно, идентификация генетических факторов, которые могут нарушить молекулярные процессы в ювенильном БАС, приобретает все большее терапевтическое значение.
7. Достижения в области секвенирования следующего поколения и клинического применения при заболеваниях двигательных нейронов у детей
Заболевания двигательных нейронов у детей часто наследуются доминантно или рецессивно, поэтому обычно проводится диагностическое генетическое тестирование. Первым направлением исследования для ребенка или молодого взрослого пациента с подозрением на проксимальную СМА должно быть тестирование MLPA на гомозиготную делецию экзонов 7 и 8 в гене SMN1 .Помимо заболеваний, связанных с SMN, заболевания мотонейронов у детей демонстрируют генетическую и фенотипическую гетерогенность, что затрудняет диагностику, основанную только на клиническом обследовании или только гистологических исследованиях. Следовательно, последовательное тестирование генов может породить дорогостоящую, долгую и неудовлетворительную диагностическую одиссею. Тем не менее, установление молекулярно-генетического диагноза важно для понимания прогноза, точного генетического консультирования, обеспечения доступа к конкретным методам лечения и клиническим испытаниям. С появлением NGS в диагностической парадигме произошла революция в области генетических заболеваний, с возможностью захвата и секвенирования ряда генов, всего экзома или генома.Эти инновационные технологии продемонстрировали эффективность в нескольких группах пациентов с нервно-мышечными расстройствами, улучшая диагностические показатели, сокращая время и стоимость диагностики и расширяя фенотипы [144, 145]. Кроме того, NGS служит мощным инструментом для обнаружения новых путей развития болезни, понимания общих путей развития болезни и определения новых целей для лечения. AANEM считает, что проведение генетического тестирования нервно-мышечных пациентов важно для предоставления высококачественной медицинской помощи нервно-мышечным пациентам с клиническими, безопасными, психосоциальными и исследовательскими преимуществами, поощряя обновление клинической практики [146].Следует отметить, что этот подход отличается от БДН у взрослых; Недавние европейские руководства рекомендовали генетическое тестирование у пациентов с родственниками первой или второй степени родства с БДН или ЛТД с информированного согласия и консультации [147]. Последнее включает понимание потенциальных неопределенностей относительно патогенности и пенетрантности некоторых генетических мутаций и недостаточных корреляций генотип-фенотип. Поскольку технологии NGS все чаще используются в качестве генетического теста первого уровня в клинической сфере, точная клиническая оценка продолжает оставаться важной.Таким образом, понимание NGS и его применения для педиатрических пациентов с подозрением на генетические нарушения двигательных нейронов является обязательным и резюмируется ниже.
NGS включает в себя целевые панели, WES и WGS. Целевые NGS, включающие панель известных генов, коммерчески доступны и ценны для диагностики в условиях с гетерогенностью и клинической вариабельностью генетических локусов [148]. Генные панели — это быстрый, относительно экономичный и комплексный тест для диагностики или исключения определенных состояний.WES анализирует кодирующую область белка, в которой, по оценкам, происходит около 85% болезнетворных мутаций. Ограничения текущих целевых панелей и WES включают в себя то, что они не могут надежно обнаруживать варианты количества копий и тройные повторяющиеся изменения. Например, количество копий SMN1 и повторяющиеся последовательности будут пропущены, что приведет к ложноотрицательным результатам. В будущих итерациях по повышению эффективности целевых подходов NGS может быть включен анализ количества копий. Дальнейшие проблемы включают большое количество вариантов интерпретации и отчетности, а также вариант с неопределенной значимостью (VOUS).Трудно провести различие между индивидуумами и редкими и непатогенными вариантами по сравнению с вариантами, вызывающими болезнь; Следовательно, существуют жизненно важные для этого валидационные тесты, такие как предсказания in silico, функциональные исследования, сложный анализ сегрегации у других членов семьи и отсутствие мутации в контролируемой популяции. Даже с этими инструментами может быть слишком много вариантов для дальнейшего изучения, особенно при работе с небольшими семьями или изолированными пробандами. К этим заболеваниям относятся те, которые настолько редки, что требуется выявление дополнительных семей во всем мире.Уровень ложных срабатываний для NGS, особенно для вставок и удалений, все еще очень высок. Секвенирование по Сэнгеру неизбежно и необходимо для устранения ложных срабатываний, но увеличивает стоимость и время обработки. Существуют стандартные конвейеры для обработки данных секвенирования, генерируемых WES, и их можно сравнивать с различными общедоступными базами данных, такими как база данных однонуклеотидного полиморфизма (SNP) (dbSNP), проект 1000 Genomes, сервер Exome Variant Server и Международный проект HapMap, а также базы данных внутреннего контроля для удаления распространенных вариантов и помощи в идентификации причинных генов.Основным ограничением WES является то, что патогенные мутации могут происходить вне экзонов или быть следствием вариантов с малым числом копий, которые можно будет идентифицировать только с помощью WGS. Его все чаще используют у недиагностированных пациентов, у которых нет обнаруживаемой геномной кодирующей мутации. WGS позволяет исследовать однонуклеотидные варианты, инделки, структурные варианты и варианты числа копий как в 1% части генома, которая кодирует последовательности белка, так и в 99% остальных некодирующих последовательностей.Зонды WGS могут также включать зонды митохондриального генома. Основная трудность для WGS заключается в том, что он может идентифицировать гораздо больше полиморфизмов последовательностей, не связанных с заболеванием. В настоящее время средства для фильтрации вариантов ограничены и все еще находятся в стадии разработки.
Дефицит гипоксантин-гуанинфосфорибозилтрансферазы (HPRT): синдром Леша-Найхана | Orphanet Journal of Rare Diseases
Lesch M, Nyhan WL: Семейное расстройство метаболизма мочевой кислоты и функции центральной нервной системы.Am J Med. 1964, 36: 561-570. 10.1016 / 0002-9343 (64)
-4.
CAS
Статья
PubMed
Google Scholar
Seegmiller JE, Rosenbloom FM, Kelley WN: ферментный дефект, связанный с связанным с полом неврологическим расстройством человека и чрезмерным синтезом пуринов. Наука. 1967, 155: 1682-1684. 10.1126 / science.155.3770.1682.
CAS
Статья
PubMed
Google Scholar
Kelley WN, Rosenbloom FM, Henderson JF, Seegmiller JE: специфический ферментный дефект при подагре, связанный с перепроизводством мочевой кислоты. Proc Natl Acad Sci USA. 1967, 57: 1735-1739. 10.1073 / pnas.57.6.1735.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Kelley WN, Greene ML, Rosenbloom FM, Henderson JF, Seegmiller JE: Дефицит гипоксантин-гуанинфосфорибозилтрансферазы при подагре. Ann Intern Med.1969, 70: 155-206.
CAS
Статья
PubMed
Google Scholar
Кател В., Шмидт Дж .: О семейном подагрическом диатезе, связанном с церебральными и почечными симптомами у маленького ребенка. Dtsch Med Wochenschr. 1959, 84: 2145-2147.
CAS
Статья
PubMed
Google Scholar
Jinnah HA, Visser JE, Harris JC, Verdu A, Larovere L, Ceballos-Picot I, Gonzalez-Alegre P, Neychev V, Torres RJ, Dulac O, Desguerre I, Schretlen DJ, Robey KL, Barabas G, Bloem BR, Nyhan W, De Kremer R, Eddey GE, Puig JG, Reich SG, Международная исследовательская группа по болезни Леша-Найхана: определение моторного расстройства при болезни Леша-Найхана.Мозг. 2006, 129: 1201-1217. 10.1093 / мозг / awl056.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Hoefnagel D, Andrew ED, Mireault NG, Berndt WO: Наследственный хореоатетоз, членовредительство и гиперурикемия у молодых мужчин. N Engl J Med. 1965, 273: 130-135.
CAS
Статья
PubMed
Google Scholar
Огасавара Н., Стаут Дж. Т., Гото Х, Сонта С. И., Мацумото А., Каски К. Т.: Молекулярный анализ пациенток Леш-Нихан.J Clin Invest. 1989, 84: 1024-1027.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Ринат С., Зореф-Шани Э., Бен-Нерия З., Бромберг Ю., Беккер-Коэн Р., Файнштейн С., Сперлинг О., Фришберг Ю.: Молекулярная, биохимическая и генетическая характеристика пациентки с заболеванием Леш- Болезнь Нихана. Mol Genet Metab. 2006, 87: 249-252. 10.1016 / j.ymgme.2005.09.025.
CAS
Статья
PubMed
Google Scholar
De Gregorio L, Jinnah HA, Harris JC, Nyhan WL, Schretlen DJ, Trombley LM, O’Neill JP: болезнь Леша-Найхана у женщины с клинически нормальным монозиготным близнецом. Mol Genet Metab. 2005, 85: 70-77. 10.1016 / j.ymgme.2004.11.009.
CAS
Статья
PubMed
Google Scholar
Де Грегорио Л., Нихан В.Л., Серафин Э., Чамолес Н.А.: неожиданно пострадавшая пациентка из классической семьи Леш-Нихан. Mol Genet Metab. 2000, 69: 263-268.10.1006 / мг.2000.2967.
CAS
Статья
PubMed
Google Scholar
Aral B, de Saint Basile G, Al-Garawi S, Kamoun P, Ceballos-Picot I: Новая бессмысленная мутация в гене гипоксантингуанинфосфорибозилтрансферазы и неслучайная X-инактивация, вызывающая синдром Леш-Найхана у пациентки. . Hum Mutat. 1996, 7: 52-58. 10.1002 / (SICI) 1098-1004 (1996) 7: 1 <52 :: AID-HUMU7> 3.0.CO; 2-R.
CAS
Статья
PubMed
Google Scholar
Хара К., Касивамата С., Огасавара Н., Охиси Х., Нацумэ Р., Яманака Т., Хакамада С., Миядзаки С., Ватанабе К.: женский случай синдрома Лича-Нихана. Tohoku J Exp Med. 1982, 137: 275-282.
CAS
Статья
PubMed
Google Scholar
ван Богарт П., Себальос И., Дегер И., Телви Л., Камун П., Понсо Г.: Синдром Леша-Найхана у девочки. J Inherit Metab Dis. 1992, 15: 790-791. 10.1007 / BF01800022.
CAS
Статья
PubMed
Google Scholar
Юкава Т., Акадзава Х., Мияке Й., Такахаши Й., Нагао Х., Такеда Э.: пациентка с синдромом Леша-Найхана. Dev Med Child Neurol. 1992, 34: 543-546.
CAS
Статья
PubMed
Google Scholar
Crawhall JC, Henderson JF, Kelley WN: Диагностика и лечение синдрома Леша-Найхана. Pediatr Res. 1972, 6: 504-513. 10.1203 / 00006450-197205000-00004.
CAS
Статья
PubMed
Google Scholar
Джинна Х.А., Фридман Т .: Болезнь Леша-Нихана и ее варианты. Метаболические и молекулярные основы наследственного заболевания. 8-е издание. Под редакцией: Скрайвер Ч.Р., Боде А.Л., Слай В.С., Валле Д. Макгроу-Хилл, Нью-Йорк; 2000: 2537-2570.
Google Scholar
Гарсиа Пуиг Дж., Торрес Хименес Р., Матеос Ф., Рамос Т., Аркас Дж., Буньо А., О’Нил П.: Спектр дефицита гипоксантин-гуанинфосфорибозилтрансферазы (HPRT). Клинический опыт основан на 22 пациентах из 18 испанских семей.Медицина (Балт). 2001, 80: 102-112. 10.1097 / 00005792-200103000-00003.
Артикул
Google Scholar
Пейдж Т., Бакай Б., Ниссинен Э., Нихан В.Л .: Варианты гипоксантин-гуанинфосфорибозилтрансферазы: корреляция клинического фенотипа с активностью фермента. J Inherit Metab Dis. 1981, 4: 203-206. 10.1007 / BF02263652.
CAS
Статья
PubMed
Google Scholar
Schretlen DJ, Harris JC, Park KS, Jinnah HA, del Pozo NO: нейрокогнитивное функционирование при болезни Леша-Найхана и частичной гипоксантин-гуанин-фосфорибозилтрансферазной недостаточности. J Int Neuropsychol Soc. 2001, 7: 805-812. 10.1017 / S135561770177703X.
CAS
Статья
PubMed
Google Scholar
Мэтьюз В.С., Солан А., Барабас Г., Роби К.: Когнитивное функционирование при синдроме Леша-Найхана: последующее 4-летнее исследование. Dev Med Child Neurol.1999, 41: 260-262. 10.1017 / S001216229
47.
CAS
Статья
PubMed
Google Scholar
Андерсон Л.Т., Эрнст М., Дэвис С.В.: Когнитивные способности пациентов с болезнью Леша-Нихана. J Autism Dev Disord. 1992, 22: 189-203. 10.1007 / BF01058150.
CAS
Статья
PubMed
Google Scholar
Андерсон Л.Т., Эрнст М.: Самоповреждение при болезни Леша-Нихана.J Autism Dev Disord. 1994, 24: 67-81. 10.1007 / BF02172213.
CAS
Статья
PubMed
Google Scholar
Schroeder SR, Oster-Granite ML, Berkson G, Bodfish JW, Breese GR, Cataldo MF, Cook EH, Crnic LS, DeLeon I, Fisher W, Harris JC, Horner RH, Iwata B, Jinnah HA, King BH, Lauder JM, Lewis MH, Newell K, Nyhan WL, Rojahn J, Sackett GP, Sandman C, Symons F, Tessel RE, Thompson T, Wong DF: Самоповреждающее поведение: отношения ген-мозг-поведение.Ment Retard Dev Disabil Res Rev.2001, 7: 3-12. 10.1002 / 1098-2779 (200102) 7: 1 <3 :: AID-MRDD1002> 3.0.CO; 2- #.
CAS
Статья
PubMed
Google Scholar
Олсон Л., Хулихан Д.: Обзор поведенческих методов лечения, используемых при синдроме Леша-Найхана. Behav Modif. 2000, 24: 202-222. 10.1177 / 0145445500242003.
CAS
Статья
PubMed
Google Scholar
ван дер Зи С.П., Шретлен Э.Д., Монненс Л.А.: Мегалобластная анемия при синдроме Леша-Найхана. Ланцет. 1968, 1: 1427-10.1016 / S0140-6736 (68)
-3.
CAS
Статья
PubMed
Google Scholar
Беккер М.А., Ресслер Б.Дж .: Гиперурикемия и подагра. Метаболические и молекулярные основы наследственного заболевания. Под редакцией: Scriver CR, Beaudet AL, Sly WS, Valle D. New York, McGraw-Hill; 1995: 1192-7
Google Scholar
Rosenbloom FM, Henderson JF, Caldwell IC, Kelley WN, Seegmiller JE: Биохимические основы ускоренного биосинтеза пурина de novo в человеческих фибробластах, лишенных гипоксантин-гуанинфосфорибозилтрансферазы. J Biol Chem. 1968, 243: 1166-1173.
CAS
PubMed
Google Scholar
Visser JE, Bar PR, Jinnah HA: болезнь Леша-Найхана и базальные ганглии. Brain Res Rev.2000, 32: 449-475. 10.1016 / S0165-0173 (99) 00094-6.
CAS
Статья
PubMed
Google Scholar
Nyhan WL: Функция дофамина при болезни Леша-Найхана. Перспектива здоровья окружающей среды. 2000, 108 (Дополнение 3): 409-411. 10.2307 / 3454529.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Watts RW, Spellacy E, Gibbs DA, Allsop J, McKeran RO, Slavin GE: Клинические, посмертные, биохимические и терапевтические наблюдения за синдромом Леша-Нихана с особым упором на неврологические проявления.Q J Med. 1982, 51 (201): 43-78.
CAS
PubMed
Google Scholar
LLoyd KG, Hornykiewicz O, Davidson L, Shannak K, Farley I, Goldstein M, Shibuya M, Kelley WN, Fox IH: Биохимические доказательства дисфункции нейротрансмиттеров головного мозга при синдроме Леш-Найхана. N Engl J Med. 1981, 305: 1106-1111.
CAS
Статья
PubMed
Google Scholar
Сильверштейн Ф.С., Джонстон М.В., Хатчинсон Р.Дж., Эдвардс Н.Л .: Синдром Леша-Найхана: аномалии нейротрансмиттеров спинномозговой жидкости. Неврология. 1985, 35: 907-911.
CAS
Статья
PubMed
Google Scholar
Jankovic J, Caskey CT, Stout JT, Butler IJ: Синдром Леша Найхана: исследование моторного поведения и нейротрансмиттеров спинномозговой жидкости. Энн Нейрол. 1988, 23: 466-469. 10.1002 / ana.410230507.
CAS
Статья
PubMed
Google Scholar
Эрнст М., Заметкин А.Дж., Маточик Дж.А., Паскуальвака Д., Джонс PH, Харди С., Ханкерсон Дж. Г., Дуде Д. Д., Коэн Р. М.: Пресинаптический дофаминергический дефицит при болезни Леша-Нихана. New Engl J Med. 1996, 334: 1568-1572. 10.1056 / NEJM199606133342403.
CAS
Статья
PubMed
Google Scholar
Wong DF, Harris JC, Naidu S, Yokoi F, Marenco S, Dannals RF, Ravert HT, Yaster M, Evans A, Rousset O, Bryan RN, Gjedde A, Kuhar MJ, Breese GR: переносчики дофамина заметно снижаются при болезни Леша-Нихана in vivo.Proc Natl Acad Sci USA. 1996, 93: 5539-5543. 10.1073 / pnas.93.11.5539.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Бриз Г.Р., Крисуэлл Х.Э., Дункан Г.Е., Мюллер Р.А.: Модель дефицита дофамина при болезни Леша-Найхана — крыса с поражением 6-OHDA у новорожденных. Brain Res Bull. 1990, 25: 477-484. 10.1016 / 0361-9230 (90)
-З.
CAS
Статья
PubMed
Google Scholar
Jinnah HA, Wojcik BE, Hunt M, Narang N, Lee KY, Goldstein M, Wamsley JK, Langlais PJ, Friedmann T: дефицит дофамина в генетической мышиной модели болезни Леша-Нихана. J Neurosci. 1994, 14: 1164-1175.
CAS
PubMed
Google Scholar
Jinnah HA, Jones MD, Wojcik BE, Rothstein JD, Hess EJ, Friedmann T., Breese GR: Влияние возраста и напряжения на потерю дофамина в полосатом теле в генетической мышиной модели болезни Леша-Нихана.J Neurochem. 1999, 72: 225-229. 10.1046 / j.1471-4159.1999.0720225.x.
CAS
Статья
PubMed
Google Scholar
Engle SJ, Womer DE, Davies PM, Boivin G, Sahota A, Simmonds HA, Stambrook PJ, Tischfield JA: Мыши с дефицитом HPRT-APRT не являются моделью синдрома Леша-Найхана. Hum Mol Genet. 1996, 5: 1607-1610. 10.1093 / hmg / 5.10.1607.
CAS
Статья
PubMed
Google Scholar
Смит Д.В., Фридман Т. Характеристика дофаминового дефекта в первичных культурах дофаминергических нейронов мышей с нокаутом по гипоксантинфосфорибозилтрансферазе. Mol Ther. 2000, 1: 486-491. 10.1006 / mthe.2000.0057.
CAS
Статья
PubMed
Google Scholar
Pelled D, Sperling O, Zoref-Shani E: Аномальное содержание пуриновых и пиримидиновых нуклеотидов в первичных культурах астроглии от трансгенных мышей с дефицитом гипоксантин-гуанинфосфорибозилтрансферазы.J Neurochem. 1999, 72: 1139-1145. 10.1046 / j.1471-4159.1999.0721139.x.
CAS
Статья
PubMed
Google Scholar
Зореф-Шани Е., Бур П., Брош С., Пеллед Д., Бромберг Ю., Сперлинг О: метаболизм пуриновых нуклеотидов в культивируемых нейронах и астроглии от мышей с нокаутом с дефицитом HPRT. Ital J Biochem. 2001, 50: 9-13.
CAS
PubMed
Google Scholar
Shirley TL, Lewers JC, Egami K, Majumdar A, Kelly M, Ceballos-Picot I, Seidman MM, Jinnah HA: Модель культуры нейрональной ткани человека для болезни Леша-Нихана. J Neurochem. 2007, 101: 841-853. 10.1111 / j.1471-4159.2007.04472.x.
CAS
Статья
PubMed
Google Scholar
Pinto CS, Jinnah HA, Shirley TL, Nyhan WL, Seifert R: Измененная мембранная активность NTPase в фибробластах болезни Леша-Найхана: сравнение с мышами с нокаутом HPRT и линиями клеток с дефицитом HPRT.J Neurochem. 2005, 93: 1579-1586. 10.1111 / j.1471-4159.2005.03151.x.
CAS
Статья
PubMed
Google Scholar
Крисвелл Х., Мюллер Р.А., Бриз Г.Р .: Оценка взаимодействий пурин-дофамин у пораженных 6-гидроксидофамином крыс: доказательства пре- и постсинаптического влияния аденозина. J Pharmacol Exp Ther. 1988, 244: 493-500.
CAS
PubMed
Google Scholar
Torres RJ, Deantonio I, Prior C, Puig JG: Транспорт аденозина в лимфоцитах периферической крови от пациентов Lesch-Nyhan. Biochem J. 2004, 377: 733-739. 10.1042 / BJ20031035.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Бертелли М., Чекчин С., Лапуччи С., Джакомелли Г., Джинна Х.А., Пандольфо М., Мичели В. Изучение аденозинергической системы в мозге мыши с нокаутом HPRT (болезнь Леша-Нихана).Clin Chim Acta. 2006, 373: 104-107. 10.1016 / j.cca.2006.05.013.
CAS
Статья
PubMed
Google Scholar
Prior C, Torres RJ, Puig JG: Гипоксантин снижает равновесный тип транспорта аденозина в лимфоцитах пациентов Lesch-Nyhan. Eur J Clin Invest. 2007, 37: 905-911. 10.1111 / j.1365-2362.2007.01869.x.
CAS
Статья
PubMed
Google Scholar
Bavaresco CS, Chiarani F, Wannmacher CM, Netto CA, Wyse AT: Интрастриатальный гипоксантин снижает активность Na (+), K (+) — АТФазы и вызывает окислительный стресс у крыс. Metab Brain Dis. 2007, 22: 1-11. 10.1007 / s11011-006-9037-л.
CAS
Статья
PubMed
Google Scholar
Pinto CS, Seifert R: Снижение GTP-стимулированной активности аденилатциклазы в HPRT-дефицитных человеческих и мышиных фибробластах и клеточных мембранах нейробластомы крысы B103.J Neurochem. 2006, 96: 454-459. 10.1111 / j.1471-4159.2005.03570.x.
CAS
Статья
PubMed
Google Scholar
МакКеран Р.О .: Факторы патогенеза поражения головного мозга и анемии при синдроме Леша-Найхана. Ciba Found Symp. 1977, 48: 83-96.
CAS
PubMed
Google Scholar
Харрис Дж. К., Ли Р. Р., Джинна Х. А., Вонг Д. Ф., Ястер М., Брайан Р. Н.: Измерение черепно-мозговой магнитно-резонансной томографии и результаты при синдроме Леш-Нихана.Arch Neurol. 1998, 55: 547-553. 10.1001 / archneur.55.4.547.
CAS
Статья
PubMed
Google Scholar
Кауфман Дж. М., Грин М. Л., Зигмиллер Дж. Э .: Соотношение мочевой кислоты и креатинина в моче — скрининговый тест для выявления наследственных нарушений пуринового обмена. Дефицит фосфорибозилтрансферазы (PRT) при Х-сцепленном церебральном параличе и подагре. J Pediatr. 1968, 73: 583-592. 10.1016 / S0022-3476 (68) 80274-4.
CAS
Статья
PubMed
Google Scholar
Torres RJ, Prior C, Puig JG: Эффективность и безопасность аллопуринола у пациентов с дефицитом гипоксантин-гуанинфосфорибозилтрансферазы. Обмен веществ. 2007, 56: 1179-1186. 10.1016 / j.metabol.2007.04.013.
CAS
Статья
PubMed
Google Scholar
Rylance HJ, Wallace RC, Nuki G: анализ гипоксантин-гуанинфосфорибозилтрансферазы с использованием высокоэффективной жидкостной хроматографии. Clin Chim Acta. 1982, 121: h259-165.10.1016 / 0009-8981 (82)
- -7.
Артикул
Google Scholar
Пуиг Дж. Г., Торрес Р. Дж., Матеос Ф. А., Рамос Т., Буно А. С., Аркас Дж.: Спектр дефицита гипоксантин-гуанинфосфорибозилтрансферазы (HPRT). Adv Exp Med Bio. 2000, 486: 15-21.
CAS
Статья
Google Scholar
Эдвардс А., Восс Х., Райс П., Чивителло А., Стегеманн Дж., Швагер С., Циммерманн Дж., Эрфле Х., Каски CT, Ансорге В. Автоматизированное секвенирование ДНК локуса HPRT человека.Геномика. 1990, 6: 593-608. 10.1016 / 0888-7543 (90)
CAS
Статья
PubMed
Google Scholar
Jolly DJ, Okayama H, Berg P, Esty AC, Filpula D, Bohlen P, Johnson GG, Shively JE, Hunkapillar T, Friedmann T: Выделение и характеристика полноразмерной экспрессируемой кДНК для человеческого гипоксантина фосфорибозила. трансфераза. Proc Natl Acad Sci U S. A. 1983, 80: 477-481. 10.1073 / pnas.80.2.477.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Wilson JM, Stout JT, Palella TD, Davidson BL, Kelley WN, Caskey CT: молекулярное исследование дефицита гипоксантин-гуанинфосфорибозилтрансферазы у человека. J Clin Invest. 1986, 77: 188-195.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Дэвидсон Б.Л., Тарле С.А., Палелла Т.Д., Келли В.Н.: Молекулярная основа дефицита гипоксантин-гуанинфосфорибозилтрансферазы у десяти субъектов, определяемая прямым секвенированием амплифицированных транскриптов.J Clin Invest. 1989, 84: 342-346.
PubMed Central
CAS
Статья
PubMed
Google Scholar
Гиббс Р.А., Нгуен П.Н., Эдвардс А., Чивителло А.Б., Каски СТ: обнаружение множественных делеций ДНК и секвенирование экзонов гена гипоксантинфосфорибозилтрансферазы в семьях Леш-Нихан. Геномика. 1990, 7: 235-244. 10.1016 / 0888-7543 (90)
-6.
CAS
Статья
PubMed
Google Scholar
Jinnah HA, De Gregorio L, Harris JC, Nyhan WL, O’Neill JP: Спектр наследственных мутаций, вызывающих дефицит HPRT: 75 новых случаев и обзор 196 ранее зарегистрированных случаев. Mutat Res. 2000, 463: 309-326. 10.1016 / S1383-5742 (00) 00052-1.
CAS
Статья
PubMed
Google Scholar
Торрес Р. Дж., Матеос Ф. А., Молано Дж., Гатхофф Б. С., О’Нил Дж. П., Гундель Р. М., Тромбли Л., Пуч Дж. Г.: Молекулярные основы дефицита гипоксантин-гуанин-фосфорибозилтрансферазы в тринадцати испанских семьях.Hum Mutat. 2000, 15: 383-10.1002 / (SICI) 1098-1004 (200004) 15: 4 <383 :: AID-HUMU17> 3.0.CO; 2-2.
CAS
Статья
PubMed
Google Scholar
Официальный сайт исследовательской группы по болезни Леша-Нихана. [http://www.lesch-nyhan.org]
Ринат К., Зореф-Шани Э., Бен-Нирия З., Бромберг Ю., Беккер-Коэн Р., Файнштейн С., Сперлинг О., Фришберг Ю. , биохимическая и генетическая характеристика пациентки с болезнью Леша-Найхана.Mol Genet Metab. 2006, 87: 249-252. 10.1016 / j.ymgme.2005.09.025.
CAS
Статья
PubMed
Google Scholar
De Gregorio L, Jinnah HA, Harris JC, Nyhan WL, Schretlen DJ, Trombley LM, O’Neill JP: болезнь Леша-Найхана у женщины с клинически нормальным монозиготным близнецом. Mol Genet Metab. 2005, 85: 70-77. 10.1016 / j.ymgme.2004.11.009.
CAS
Статья
PubMed
Google Scholar
De Gregorio L, Nyhan WL, Serafin E, Chamoles NA: неожиданно пострадавшая пациентка из классической семьи Lesch-Nyhan. Mol Genet Metab. 2000, 69: 263-268. 10.1006 / мг.2000.2967.
CAS
Статья
PubMed
Google Scholar
Aral B, de Saint Basile G, Al-Garawi S, Kamoun P, Ceballos-Picot I: новая бессмысленная мутация в гене гипоксантингуанинфосфорибозилтрансферазы и неслучайная X-инактивация, вызывающая синдром Леш-Нихана у пациентки. .Hum Mutat. 1996, 7: 52-58. 10.1002 / (SICI) 1098-1004 (1996) 7: 1 <52 :: AID-HUMU7> 3.0.CO; 2-R.
CAS
Статья
PubMed
Google Scholar
МакКеран Р.О., Эндрюс Т.М., Хауэлл А., Гиббс Д.А., Чинн С., Уоттс В.Е.: Диагностика состояния носительства для синдрома Леша-Найхана. Q J Med. 1975, 44: 189-205.
CAS
PubMed
Google Scholar
Пуиг Дж. Г., Матеос Ф. А., Торрес Р. Дж., Буно А. С.: Пуриновый метаболизм у гетерозигот женского пола при дефиците гипоксантин-гуанинфосфорибозилтрансферазы.Eur J Clin Invest. 1998, 28: 950-957. 10.1046 / j.1365-2362.1998.00392.x.
CAS
Статья
PubMed
Google Scholar
Hakoda M, Hirai Y, Akiyama M, Yamanaka H, Terai C, Kamatani N, Kashiwazaki S: Селекция клеток крови, дефицитных по гипоксантинфосфорибозилтрансферазе (HPRT), у гетерозиготных стволовых клеток Lesch-Nyhan происходит на уровне мультипотентных стволовых клеток. клетки. Hum Genet. 1995, 96: 674-680. 10.1007 / BF00210298.
CAS
Статья
PubMed
Google Scholar
Бакай Б., Такер-Пиан С., Сигмиллер Дж. Э .: Выявление носителей синдрома Леша-Найхана: анализ корней волос на HPRT с помощью электрофореза в агарозном геле и авторадиографии. Clin Genet. 1980, 17: 369-374.
CAS
Статья
PubMed
Google Scholar
О’Нил Дж. П.: Тестирование носителей мутаций в семьях с синдромом Леша-Найхана: частота мутантов HPRT и анализ мутаций с Т-лимфоцитами периферической крови. Genet Test. 2004, 8: 51-64.10.1089 / 10
Артикул
PubMed
Google Scholar
Торрес Р. Дж., Буно А., Матеос Ф. А., Пуч Дж. Г.: Состояние носителя при дефиците HGPRT. Исследование в 14 испанских семьях. Adv Exp Med Biol. 1998, 431: 197-200.
CAS
Статья
PubMed
Google Scholar
Nyhan WL, Vuong LU, Broock R: Пренатальная диагностика болезни Леша-Нихана. Prenat Diagn.2003, 23: 807-809. 10.1002 / pd.691.
Артикул
PubMed
Google Scholar
Graham GW, Aitken DA, Connor JM: Пренатальная диагностика с помощью анализа ферментов у 15 беременностей с риском синдрома Леша-Найхана. Prenat Diagn. 1996, 16: 647-651. 10.1002 / (SICI) 1097-0223 (199607) 16: 7 <647 :: AID-PD932> 3.0.CO; 2-S.
CAS
Статья
PubMed
Google Scholar
Rundles RW: Разработка аллопуринола. Arch Intern Med. 1985, 145: 1492-1503. 10.1001 / archinte.145.8.1492.
CAS
Статья
PubMed
Google Scholar
Christie R, Bay C, Kaufman IA, Bakay B, Borden M, Nyhan WL: Болезнь Леша-Найхана: клинический опыт с девятнадцатью пациентами. Разработка Med Child Neurol. 1982, 24: 293-306.
CAS
Статья
PubMed
Google Scholar
Kelley WN, Rosenbloom FM, Miller J, Seegmiller JE: ферментативная основа вариабельности реакции на аллопуринол. Дефицит гипоксатин-гуанинфосфорибозилтрансферазы. N Eng J Med. 1968, 278: 287-293.
CAS
Статья
Google Scholar
Брок В.А., Голден Дж., Каплан Г.В.: Ксантиновые камни при синдроме Леша-Найхана. J Urol. 1983, 130: 157-159.
CAS
PubMed
Google Scholar
Olson L, Houlihan D: Обзор поведенческих методов лечения, используемых при синдроме Леша-Найхана. Behav Modif. 2000, 24: 202-222. 10.1177 / 0145445500242003.
CAS
Статья
PubMed
Google Scholar
McManaman J, Tam DA: Габапентин для самоповреждающего поведения при синдроме Леша-Найхана. Pediatr Neurol. 1999, 20: 381-382. 10.1016 / S0887-8994 (98) 00166-0.
CAS
Статья
PubMed
Google Scholar
Dabrowski E, Smathers SA, Ralstrom CS, Nigro MA, Leleszi JP: Ботулинический токсин как новый метод лечения членовредительства при синдроме Леша-Нихана. Dev Med Child Neurol. 2005, 47: 636-639. 10.1017 / S0012162205001246.
PubMed
Google Scholar
Pralong E, Pollo C, Coubes P, Bloch J, Roulet E, Tetreault MH, Debatisse D, Villemure JG: Электрофизиологические характеристики нейронов лимбической и моторной внутренней области бледного шара (GPI) в двух случаях Lesch-Nyhan синдром.Neurophysiol Clin. 2005, 35: 168-173. 10.1016 / j.neucli.2005.12.004.
CAS
Статья
PubMed
Google Scholar
Тайра Т., Кобаяси Т., Хори Т. Исчезновение членовредительского поведения у пациента с синдромом Леш-Нихана после двусторонней хронической стимуляции внутреннего бледного шара. История болезни. J Neurosurg. 2003, 98: 414-416.
Артикул
PubMed
Google Scholar
Нейчев В.К., Джинна Х.А.: Внезапная смерть при болезни Леша-Нихана. Dev Med Child Neurol. 2006, 48: 923-926. 10.1017 / S0012162206002015.
PubMed Central
Статья
PubMed
Google Scholar
(PDF) Нейрохирургическое лечение детей с кровоточащим диатезом: Аудит неврологического исхода
Кровоточащий диатез у педиатрических нейрохирургических пациентов
J Neurosurg Pediatr Volume 21 • Январь 2018 43
Благодарности
Мы благодарим наших пациентов .Мы благодарим
доктора Филипа Мерфи и команду тологов больницы Бомонт hema-
за помощь в сборе данных.
Ссылки
1. Андерст Дж. Д., Карпентер С.Л., Абшир TC: Оценка
нарушений свертываемости крови при подозрении на жестокое обращение с детьми. Педиатрия
131: e1314 – e1322, 2013
2. Beers SR, Wisniewski SR, Garcia-Filion P, Tian Y, Hahner
T, Berger RP и др.: Срок действия педиатрической версии
Glasgow Outcome Масштаб увеличен.J Neurotrauma
29: 1126–1139, 2012
3. Бассел Дж. Б., Танли С., Петерсон Х. С.: Благоприятный неврологический исход —
приходятся на 7 случаев перинатального внутричерепного кровоизлияния из-за
иммунной тромбоцитопении. Am J Pediatr Hematol Oncol
13: 156–159, 1991
4. Карпентер С.Л., Абшир Т.С., Андерст Дж. Д.: Оценка подозреваемого
предполагаемого жестокого обращения с детьми: состояния, предрасполагающие к кровотечению.
Педиатрия 131: e1357 – e1373, 2013
5.Cermelj M, Negro F, Schijman E, Ferro AM, Acerenza M,
Pollola J: Нейрохирургическое вмешательство у ребенка с гемофилией
с субдуральной и внутримозговой гематомой. Гемофилия
10: 405–407, 2004
6. Даути Х.А., Коулз Дж., Пармар К., Баллок П., Сэвидж Г.Ф.:
успешное удаление кровоточащей внутричерепной опухоли у больного тяжелой формой гемофилии
с использованием постоянной постоянной дозы. инфу-
моноклонального фактора VIII. Свертывание крови Фибринолиз
6: 31–34, 1995
7.Гаспаретто Е.Л., Бенитес Филхо П.Р., Даваус Т., де Карвалью
Нето А: Кровоизлияние в центральную нервную систему при тромбоцитах —
пациентов с топенией: результаты компьютерной томографии в 21 случае.
Arq Neuropsiquiatr 65 (2A): 268–272, 2007
8. Джексон Дж., Карпентер С., Андерст Дж .: Проблемы в оценке возможного злоупотребления
: проявления врожденного кровотечения
расстройства в детстве. Игнорирование жестокого обращения с детьми 36: 127–134, 2012
9. Грили К.С.: Состояния, принимаемые за травму головы, у Дженни
C (ред.): Жестокое обращение с детьми и пренебрежение: диагноз, лечение
и доказательства.Сент-Луис: Elsevier Saunders, 2011, стр. 441–
450
10. Laguna P, Klukowska A: Внутричерепное кровоизлияние у мальчика
с тяжелой гемофилией A и ингибитором фактора VIII. Чайлдс
Nerv Syst 22: 432–435, 2006
11. Laposata ME, Laposata M:
Дети с признаками жестокого обращения: когда это не жестокое обращение с детьми? Am J Clin Pathol 123
(Suppl): S119 – S124, 2005
12. Ли LK, Dayan PS, Gerardi MJ, Borgialli DA, Badawy
MK, Callahan JM и др.: Внутричерепное кровоизлияние после тупой головы
травма у детей с нарушением свертываемости крови.J Pediatr
158: 1003–1008, 2011
13. Lee WS, Chong LA, Begum S, Abdullah WA, Koh MT, Lim
EJ: Ингибитор фактора V при внутричерепном кровоизлиянии новорожденных
вторичный по отношению к тяжелому врожденному фактору V ловкость. J Pediatr
Hematol Oncol 23: 244–246, 2001
14. Ljung RC: Внутричерепное кровоизлияние при гемофилии A и B.
Br J Haematol 140: 378–384, 2008
15. Mishra P, Naithani R, Долай Т., Бхаргава Р., Махапатра М.,
Диксит А. и др.: Внутричерепное кровотечение у пациентов с
генитальными гемостатическими дефектами.Гемофилия 14: 952–955, 2008
16. Майлз Л.М., Массикотт П., Дрейк Дж.: Внутричерепное кровоизлияние у
новорожденных с нераспознанной гемофилией A: постоянная проблема
lem. Pediatr Neurosurg 34:94 –97, 20 01
17. Нельсон MD младший, Мейдер М.А., Уснер Д., Митчелл В.Г., Фенстер-
Машер М.Дж., Уилсон Д.А. и др.: Распространенность и частота
внутричерепных кровоизлияний в популяция детей с
гемофилией. Исследование роста и развития гемофилии
.Гемофилия 5: 306–312, 1999
18. Патироглу Т., Оздемир М.А., Унал Э., Алтунер Торун Ю., Коскун
А, Менку А. и др.: Внутричерепное кровоизлияние у детей с
врожденными факторами. Childs Nerv Syst 27: 1963–
1966, 2011
19. Псайла Б., Петрович А., Пейдж Л.К., Менелл Дж., Шонхольц М.,
Буссель Дж. Б.: Внутричерепное кровоизлияние (ICH) у детей с иммунной тромбоцитопенией
): изучение 40 кейсов. Кровь
114: 4777–4783, 2009
20.Шапиро А: Лечение ингибиторами: современное состояние. Semin He-
matol 38 (4 Suppl 12): 26–34, 2001
21. Strahle J, Garton HJ, Maher CO, Muraszko KM, Keep RF,
Xi G: Механизмы гидроцефалии у новорожденных и взрослых
внутрижелудочковое кровоизлияние. Transl Stroke Res 3 (Suppl
1): 25–38, 2012
22. Woo D, Broderick JP: Спонтанное внутримозговое кровоизлияние —
rhage: эпидемиология и клинические проявления. Neurosurg
Clin N Am 13: 265–279, v, 2002
23.Юэ КП, Манн К.С.: Хирургическое лечение внутричерепных гематом
у детей с гемофилией. Перспективное исследование.
Childs Nerv Syst 2: 5–9, 1986
Раскрытие информации
Авторы сообщают об отсутствии конфликта интересов в отношении материалов или методов, использованных в этом исследовании, или результатов, указанных в этой статье
.
Вклад авторов
Концепция и дизайн: Закария. Сбор данных: Закария.
Анализ и интерпретация данных: Закария, Калиаперумал, Кейрд.
Составление статьи: Закария. Критически переработал статью: Kalia-
perumal, Crimmins, Caird. Проверена представленная версия скрипта manu-
: все авторы. Утвержден окончательный вариант рукописи
от имени всех авторов: Закария. Административная / техническая / материальная —
риал поддержка: Закария. Научное руководство: Crimmins.
Корреспонденция
Зайтун Закария, отделение детской нейрохирургии,
Детская университетская больница, Temple St., Дублин 1, Ирландия.
электронная почта: [email protected].
Болезнь Гоше | МедЛинк Неврология
Клинические проявления
Презентация и курс
На основании клинических признаков и симптомов болезнь Гоше была разделена на 3 подтипа: тип 1 (ненейронопатический), тип 2 (острый нейронопатический) и тип 3 (хронический нейронопатический) (74). Все 3 типа болезни Гоше вызваны дефицитом кислой бета-глюкоцереброзидазы, что приводит к накоплению глюкоцереброзида в клетках ретикулоэндотелиальной системы.Принципиальные различия между подтипами заключаются в наличии неврологических проявлений, непосредственно вызванных дефицитом глюкоцереброзидазы. Нейронопатическая болезнь Гоше определяется как наличие неврологического поражения у пациента с биохимически подтвержденной болезнью Гоше, для которой нет другого объяснения, кроме болезни Гоше (130). Поскольку симптомы всех трех различных типов могут начаться в младенчестве, а различение подтипов зависит от развития клинических проявлений, особенно поражения нервной системы, эта классификация более уместна, чем та, которая включает ссылку на возраст начала (т. Е. » взрослая, «детская» и «ювенильная» формы болезни Гоше).Определение типа, особенно у детей, должно производиться только после тщательного обследования на наличие и прогрессирование связанных неврологических аномалий или если исследование генотипа показывает наличие аллеля N370S, который обычно ассоциируется с болезнью Гоше 1 типа.
Тип 1: Хроническая ненейронопатическая болезнь Гоше. Возраст начала и тяжесть симптомов внутри этого подтипа сильно различаются и не полностью объясняются генотипом. Хотя генетический дефект присутствует с момента зачатия, диагноз часто ставится в более позднем возрасте.Вероятно, что многим пациентам с заболеванием типа 1 не ставится диагноз из-за отсутствия значимых клинических проявлений.
Хотя бывают и исключения, обычно начальными признаками являются безболезненная спленомегалия с тромбоцитопенией, анемией и лейкопенией. У многих пациентов эти осложнения не опасны для жизни и могут оставаться незамеченными в течение многих лет.
Висцеральные элементы . Хотя гепатомегалия часто отмечается в то время, когда наблюдается спленомегалия, печень может увеличиваться только на более поздних этапах развития болезни.Умеренная печеночная дисфункция определяется по повышению уровня печеночных ферментов в сыворотке крови. Гистологически у всех пациентов имеется определенная степень фиброза печени (66). Печеночная недостаточность может возникнуть у нелеченных пациентов. Портальная гипертензия, приводящая к варикозному расширению вен пищевода, является известным осложнением, возникающим у пациентов с тяжелым заболеванием типа 1 и 3.
Гепатоспленомегалия при болезни Гоше
Массивная гепатоспленомегалия у пациента с болезнью Гоше 1 типа. (Предоставлено доктором.Джон Барранджер.)
Накопление кислой бета-глюкоцереброзидазы при болезни Гоше
Накопление глюкоцереброзидазы в селезенке приводит к образованию фиброзных рубцов и узелков. (Предоставлено доктором Джоном Барранджером.)
Печеночная ткань при болезни Гоше
Обратите внимание на сохраненный субстрат в синусоидах печени. (Предоставлено доктором Джоном Барранджером.)
Гематологические особенности . У пациентов может быть количество тромбоцитов ниже 50 000 без сопутствующего кровоточащего диатеза.И наоборот, некоторые пациенты с болезнью Гоше, у которых нормальное протромбиновое время, частичное тромбопластиновое время и количество тромбоцитов (более 100000) могут иметь ненормальное время свертывания, чрезмерное образование синяков и неожиданное периоперационное кровотечение. Эта вариабельность гематологических характеристик не дает практических рекомендаций практикующему врачу и требует индивидуального подхода, который может потребовать введения тромбоцитов и свежезамороженной плазмы до и после хирургических процедур.
Особенности скелета .Дегенеративные изменения скелета являются основной причиной инвалидности у пациентов с заболеванием 1 типа. Некоторая степень остеопении и остеолиза наблюдается практически у всех пациентов. Однако степень поражения костей варьирует (142; 65). Очень немногие пациенты не имеют ни рентгенологических, ни сцинтиграфических свидетельств поражения кости. Большинство пораженных людей имеют тяжелое бремя болезни костей, которая прогрессирует и приводит к клиническим проявлениям. Другие имеют настолько серьезное поражение, что в раннем возрасте они прикованы к инвалидной коляске из-за боли, патологического перелома или нестабильности скелета.Многие пациенты испытывают эпизодические боли в бедрах, ногах, спине и плечах, продолжающиеся от нескольких дней до месяцев. Эти эпизоды получили название «костные кризы».
Костные аномалии при болезни Гоше
Обнаружены аномалии костей в тазобедренном суставе. (Предоставлено доктором Джоном Барранджером.)
Деформация колбы Эрленмейера
Деформация колбы Эрленмейера — обычная черта пациентов с болезнью Гоше. (Предоставлено доктором.Джон Барранджер.)
Легочная гипертензия . Легочная гипертензия — относительно частое осложнение, обнаруживаемое примерно у 30% пациентов с болезнью Гоше (Mistry, личное сообщение). К счастью, в большинстве случаев это легкие и субклинические случаи с давлением в легочной артерии ниже 25 мм рт. Менее чем у 1% пациентов с известной болезнью Гоше легочная гипертензия клинически очевидна.Согласно имеющимся данным, это осложнение является прямым следствием накопления кислой бета-глюкоцереброзидазы и усугубляется у пациентов с удаленной спленэктомией из-за накопления накопительных клеток в легких. В тяжелых случаях легочной гипертензии действительно наблюдается положительное улучшение ферментной терапии (78).
Большинство пациентов с заболеванием типа 1 не имеют клинических симптомов поражения сердца или легких. Небольшое количество пациентов с заболеваниями типа 1 и 3 имеют тяжелую хроническую болезнь легких без поражения сердца.У них гипоксия, цианоз и клубнирование вследствие шунтирования, а также обширное заболевание печени.
Офтальмологические отклонения . Они редки, но изменчивы и включают проявления, затрагивающие стекловидное тело, сетчатку, роговицу, сосудистую оболочку и конъюнктиву (35). Помутнения стекловидного тела являются наиболее распространенными и наблюдаются в основном у пациентов с 3 типом Гоше и иногда требуют витрэктомии для улучшения остроты зрения.
Тип 2: Острая нейронопатическая болезнь Гоше. В отличие от вариабельности, наблюдаемой при болезни Гоше 1-го типа, из обширного обзора случаев 2-го типа, проведенного Фредриксоном, и из нашего опыта с более чем 15 случаями, очевидно, что болезнь Гоше 2-го типа является несколько более однородной по своей форме.Не имеет этнических пристрастий (45). Средний возраст дебюта составляет 3 месяца, и признаком этого обычно является массивная гепатоспленомегалия.
Неврологические отклонения . Неврологические осложнения развиваются в возрасте от 3 до 6 месяцев. Присутствующие признаки обычно указывают на вовлечение ядер черепных нервов и экстрапирамидного тракта. Классическая триада — тризм, косоглазие, сходящееся косоглазие из-за двустороннего паралича 6-го нерва с последующим завершенным экстраокулярным параличом и ретрофлексией головы сочетается с типичной лицевой дистонической позой у большинства пациентов, сопровождающейся прогрессирующей спастичностью, гиперрефлексией, положительным результатом Бабинского. признаки и другие патологические рефлексы.Развиваются дисфагия и трудности с обработкой секретов, часто за которыми следует аспирационная пневмония. Возможны судороги. По мере развития неврологического ухудшения ребенок обычно становится апатичным и неподвижным. Смерть наступает либо от апноэ, либо от аспирационной пневмонии в среднем в возрасте 9 месяцев, с диапазоном от 1 месяца до 2 лет.
Пациент с болезнью Гоше 2 типа
У этого пациента с болезнью Гоше 2 типа обратите внимание на массивную гепатоспленомегалию, кахексию, косоглазие, корковые большие пальцы и ретрофлексию шеи.(Предоставлено доктором Джоном Барранджером.)
Эта форма болезни Гоше проявляет изменения в ультраструктуре эпидермиса, которые могут служить ранним и специфическим диагностическим инструментом (24).
Болезнь Гоше новорожденных. Редкая и уникальная форма болезни Гоше проявляется водянкой плода и коллодием кожи (125). В обширном молекулярном анализе 31 пациента с этим типом врожденной болезни Гоше гомозиготность по рекомбинантному аллелю, включающему мутацию p.Leu483Pro (ранее Leu444Pro) был связан с ранней летальностью (120).
Следует еще раз подчеркнуть, что дети с болезнью Гоше 1 типа также диагностируются в возрасте до 2 лет. Они могут быстро прогрессировать со стороны костей, печени и селезенки. Некоторым из этих случаев был ошибочно поставлен диагноз типа 2. Важно, чтобы вовлечение центральной нервной системы было задокументировано и установлено как связанный результат до постановки диагноза заболевания типа 2.
Болезнь Гоше, неонатальная форма
На этой фотографии изображен пациент с неонатальной болезнью Гоше.(Предоставлено доктором Джоном Барранджером.)
Опубликован обширный обзор клинических и гематологических особенностей неонатальной болезни Гоше (86). Из этого исследования можно сделать вывод, что неонатальная болезнь Гоше диагностируется на основании патологических данных патологоанатомического исследования, при этом семейный анамнез подозревается в 22%.
В отличие от того, что наблюдается при болезни типа 2, у новорожденных с болезнью Гоше наблюдается 22% -ная частота дисморфических особенностей, характеризующихся низко посаженными ушами, маленьким носом с плоской переносицей и антевертированными ноздрями.
Характеристики клинического фенотипа и беременности несколько различаются между новорожденными и плодами, пораженными неиммунной водянкой плода, у которых недоношенность, гибель плода и неонатальный дистресс являются характерными чертами. Гепатоспленомегалия подозревается пренатально в большинстве случаев неиммунной водянки плода. Кардиомегалия также наблюдалась у некоторых плодов с болезнью Гоше, связанной с неиммунной водянкой плода (86).
Тип 3: подострая или хроническая нейронопатическая болезнь Гоше. Клинические признаки болезни Гоше 3-го типа, помимо тех, которые относятся к нервной системе, являются общими для болезни Гоше 1-го типа. Гепатоспленомегалия часто является характерным признаком, а не неврологическими аномалиями.
В хорошо задокументированных и биохимически подтвержденных случаях наблюдаются заметные различия в возрасте начала и тяжести поражения органов.
Неврологические отклонения . Неврологическим признаком болезни Гоше 3-го (и 2-го) типа является горизонтальный надъядерный паралич взора.Он присутствует у всех пациентов с нейронопатической болезнью Гоше (130; 15). Помимо медленных саккад, у этих пациентов были выявлены другие глазодвигательные аномалии, включая снижение прироста плавного преследования и вестибулоокулярного рефлекса (21). Другие проявления включают умственную отсталость различной степени, атаксию, дистонию (особенно затылочную и лицевую) и, реже, большие или психомоторные судороги и слабоумие. Реже возникает синдром прогрессирующей миоклонической эпилепсии, деменции и различной степени надъядерной офтальмоплегии с относительно легким системным заболеванием.Определенные мутации GBA1 связаны с миоклонической энцефалопатией (например, N188S), тогда как L444P / L444P редко, если вообще когда-либо, является (99; 75). Существуют значительные различия в неврологической картине у разных этнических групп. Уникальная оппозиционная поведенческая аномалия наблюдается у египетских пациентов с типом Гоше 3 (02).
В целом, пациенты могут иметь любую комбинацию системных и неврологических аномалий, включая легкое системное заболевание с прогрессирующим неврологическим течением или лежащие в основе тяжелые скелетные и гематологические аномалии с непрогрессирующим неврологическим течением и с преобладанием горизонтального надъядерного паралича взгляда (15).У пациентов, которые в остальном стабильны с медицинской и неврологической точки зрения, которые обычно имеют фенотип p.Leu483Pro / p.Leu483Pro (ранее гомозиготный L444P) и которые находятся на длительной заместительной ферментной терапии, могут развить частичные комплексные приступы височной доли прогрессирующая миоклоническая энцефалопатия (06; 99). Внезапная неожиданная смерть, часто сопровождающаяся эпилепсией, наблюдается редко, но особенно часто у пациентов из Египта (01).
Группа пациентов с нейродегенеративным течением и кальцификацией клапанов сердца и стенозом гомозиготна по мутации в гене кислой бета-глюкоцереброзидазы (GBA1) (стр.Asp409His, теперь аннотированный как NM_000157.3: c.1342G> C; p.Asp448His) (27; 76). Эти случаи были классифицированы как тип 3c. Неврологические особенности возникают в более старшем возрасте и не отличаются от типичного 3 типа болезни Гоше. Наконец, в одном отчете описан нейронопатический вариант, характеризующийся гидроцефалией, помутнением роговицы, деформацией пальцев ног и фиброзным утолщением капсул селезенки и печени у пациента с 1 п. Аллель Asp409His (64). Начало проявления в 4 месяца и своеобразная симптоматика отличают этот вариант от типа 3c и более точно имитируют болезнь Гоше 2 типа.Было опубликовано описание новых пациентов и подробный обзор литературы (76). Патогенез сердечно-сосудистых аномалий в этом гомозиготном генотипе неизвестен, но, вероятно, связан не с недостаточностью кислой бета-глюкозидазы как таковой, а скорее с другими функциями или взаимодействиями этого белка.
Клинические особенности нейронопатических фенотипов болезни Гоше, по-видимому, являются частью континуума тяжести заболевания, как описано Goker-Alpan на ряде пациентов, которые имели промежуточные фенотипы между заболеваниями типа 2 и типа 3 (54).Промежуточный фенотип, т.е. представляющий характеристики типа 2 и типа 3 с ассоциированными дисморфическими особенностями, также был описан у пациента из Пакистана (16).
Гоше и паркинсонизм. За последние несколько лет стало ясно, что болезнь Паркинсона или другие формы паркинсонизма (синуклеинопатии) возникают чаще, чем ожидалось случайно, у пациентов с болезнью Гоше, но также и у носителей мутаций GBA (108). Было обнаружено, что пациенты с болезнью Гоше и паркинсонизмом имеют типичные тельца Леви в областях гиппокампа, восприимчивых к нейродегенерации, связанной с Гоше, что патологически подтверждает совместное возникновение патологий Гоше и Паркинсона (143).Большое метааналитическое исследование убедительно продемонстрировало, что отношение шансов для любой мутации GBA1 у пациентов по сравнению с контрольной в целом составляло 5,43 (116). Пациенты с мутацией GBA1 , выявленные ранее с этим заболеванием, с большей вероятностью имели пораженных родственников и с большей вероятностью имели атипичные клинические проявления. Таким образом, мутации GBA1 и являются крупнейшим генетическим фактором риска болезни Паркинсона (116). Важно отметить, что пациенты с типом Гоше 1 также подвержены более высокому риску (22).Механизм, по которому мутации GBA1 являются фактором риска паркинсонизма, неясен, но поскольку существует четкая взаимосвязь между серьезностью мутации и вероятностью и возрастом начала развития болезни Паркинсона, вероятно, что потеря задействована функция кислой бета-глюкоцереброзидазы (49). Действительно, активность глюкоцереброзидазы, измеренная в периферической крови, оказалась ниже у пациентов с болезнью Паркинсона, даже если они не несут мутации GBA1 (05).Дефицит глюкоцереброзидазы был также обнаружен в черной субстанции головного мозга при болезни Паркинсона, не имеющей мутаций GBA1 (52). Эти данные были подтверждены в другой группе, которая идентифицировала избирательное снижение уровня глюкоцереброзидазы у пациентов с болезнью Паркинсона и диффузной болезнью тельцов Леви, предполагая возможную роль глюкоцереброзидазы как биомаркера синуклеинопатии (26). Полученные данные подтверждают, что дефицит глюкоцереброзидазы и альфа-синуклеина образуют петлю положительной обратной связи, которая может привести к самораспространяющейся болезни, которая в конечном итоге приводит к гибели нейронов (85).Это объяснение дополнительно подтверждается обнаружением дефицита глюкоцереброзидазы в черной субстанции мозга при болезни Паркинсона, не имеющей мутаций GBA1 (52). Противоречивое объяснение состоит в том, что мутированная глюкоцереброзидаза вызывает стресс эндоплазматического ретикулума в восприимчивых нейронах (82). Эти 2 механизма могут действовать согласованно, но в любом случае мутации GBA1 и являются наиболее распространенным генетическим фактором риска синуклеинопатий (болезни Паркинсона и деменции с тельцами Леви).Проспективное исследование пациентов с болезнью Гоше и носителей мутации GBA1 показало, что клинические признаки, связанные с премоторной болезнью Паркинсона, и двигательные особенности болезни Паркинсона более распространены по сравнению с контрольной группой и прогрессируют в течение 2-летнего наблюдения (14). Эти данные подтверждают гипотезу о том, что у некоторых людей с положительной мутацией GBA1 и проявляются клинические признаки ранней нейродегенерации (14). Однако дефицит других лизосомальных ферментов может быть факторами риска развития болезни Паркинсона (106), хотя не было обнаружено снижения этих активностей в крови носителей и пациентов с болезнью Паркинсона, не являющихся носителями (05).
Прогноз и осложнения
Смерть у пациентов типа 1 по Гоше, которые никогда не получали заместительную ферментную терапию, наступила в среднем в возрасте 66 лет и часто была вызвана заболеванием печени, сепсисом, злокачественными новообразованиями и самоубийством (138).
Вовлечение внутренних органов.
Селезенка . Безболезненная спленомегалия обычно является самым ранним признаком всех типов болезни Гоше. Даже если селезенка не пальпируется при физикальном обследовании, обычно можно продемонстрировать ее увеличение с помощью методов диагностической визуализации.Скорость увеличения селезенки помогает судить о скорости прогрессирования заболевания. Предпочтительным методом измерения объема органа является объемная МРТ (25). Скорость увеличения селезенки часто одинакова для каждого случая, но известно, что изменения скорости прогрессирования происходят без каких-либо провоцирующих факторов. Изменения в этой частоте были связаны со злокачественными новообразованиями или другими интеркуррентными заболеваниями. Самопроизвольный разрыв селезенки случается редко. В большинстве случаев развивается гиперспленизм, проявляющийся панцитопенией и кровоточащим диатезом.Время выживания эритроцитов и тромбоцитов сокращается. Спленэктомия должна быть ограничена пациентами с тяжелым кровоточащим диатезом, сердечной недостаточностью с высоким выбросом или механическим поражением кишечника, диафрагмы или почек. Ткань селезенки широко изучена. Обычно наблюдается фиброз с искажением архитектуры селезенки. Селезенка увеличена и тверда и содержит бледные участки, вызванные инфарктами. Процесс инфаркта может привести к боли в животе и должен учитываться при дифференциальной диагностике острого живота у пациентов с болезнью Гоше.Красная пульпа селезенки заменяется белыми скоплениями клеток Гоше. На поверхности и теле селезенки могут быть темно-пурпурные узелки, являющиеся очагами экстрамедуллярного кроветворения. Спленэктомия почти всегда сопровождается коррекцией сердечных и гематологических нарушений. Однако спленэктомия представляет собой риск тяжелого тяжелого сепсиса, который может привести к летальному исходу в течение дня. По этим причинам заместительная ферментная терапия является методом выбора.
Печень . Несмотря на частое возникновение гепатомегалии при болезни Гоше, печеночная недостаточность возникает нечасто.Клетки Гоше видны внутри синусоид. Практически во всех случаях возникает фиброз. В более тяжелых случаях фиброз искажает архитектуру, образуя небольшие регенерирующие узелки, которые инфильтрируются клетками Гоше. Эти случаи были описаны как цирротические. В отличие от других липидозов (таких как болезнь Ниманна-Пика), гепатоциты не участвуют в хранении. Увеличенная печень может содержать участки экстрамедуллярного кроветворения. У большинства пациентов наблюдаются отклонения от нормы функциональных тестов печени.Заметная портальная гипертензия и последующие осложнения, такие как асцит и варикозное расширение вен пищевода, действительно возникают и являются обычным явлением при тяжелом заболевании. Радионуклидное сканирование в большинстве случаев показывает смещение индикатора от печени к селезенке, что указывает на степень портальной гипертензии. Рецидивирующее кровотечение из варикозно расширенных вен пищевода успешно лечится с помощью комбинации агрессивного медикаментозного лечения и склеротерапии. Желтуха является серьезным признаком этого заболевания и представляет собой интеркуррентный инфекционный, хронический активный гепатит или печеночную недостаточность.По опыту авторов, только 4 пациента из нескольких сотен умерли от печеночной недостаточности. В случаях поражения печени показано раннее вмешательство с заместительной ферментной терапией. Трансплантация печени требуется пациентам с терминальной стадией заболевания печени.
У некоторых пациентов лимфатические узлы могут быть увеличены и содержать клетки Гоше. Часто поражаются вилочковая железа, бляшки Пейера в кишечнике и глоточные миндалины.
Скелетные проявления. Степень тяжести и начало симптомов очень различаются.Боль в суставах и «артритная» боль в костях являются обычными. Эти боли вызваны повреждением скелета в околосуставных областях кости и купируются анальгетиками. Особенно полезны нестероидные противовоспалительные средства.
У пациентов с Гоше типа 1 и 3 часто встречаются рентгенографические аномалии костей, встречающиеся у 50–75% всех пациентов. Часто наблюдается расширение коры в дистальном отделе бедренной кости (деформация колбы Эрленмейера), а также переломы и другие аномалии вертлужной впадины, головки и шейки бедренной кости.Замена протеза тазобедренного сустава неоценима, поскольку позволяет пациентам оставаться в амбулаторных условиях. Поражения бедра в некоторых случаях можно спутать с болезнью Легга-Кальве-Пертеса. Разрушение тел позвонков может вызвать коллапс и образование гиббуса, а также дисфункцию спинного мозга или нервов.
Обширное поражение костей у пациента с болезнью Гоше
На поздних стадиях болезни часто требуется несколько ортопедических процедур. (Предоставлено доктором Джоном Баррангером.)
Боль в костях и костные кризы. Инфаркт кости вызывает «костный перелом». Этот процесс чаще поражает головки бедренной кости и дистальный отдел бедренной кости, чем другие кости. Эти эпизоды обычно самодостаточны и длятся примерно 2 недели, но могут длиться до многих месяцев. В первые дни кризиса обезболивание трудно осуществить и может потребоваться госпитализация. Постельный режим всегда показан до полного разрешения эпизода. Иногда инфарктный процесс проходит полностью, но обычно развивается область остеосклероза, деформации кости или патологического перелома.Существует вероятность того, что область инфаркта станет вторично инфицированной, что приведет к истинному остеомиелиту. Однако это не является однородным, и эпизод следует лечить консервативно, если нет серьезных подозрений на инфекцию. Следует избегать обработки инфарктом кости без четких указаний, поскольку это может привести к вторичной инфекции и развитию синусового тракта. Рентген и сканирование технеция обычно не помогают отличить инфаркт от остеомиелита, но сканирование галлия может помочь в этом, иногда трудном решении.В качестве общей меры следует убедиться, что поддерживается адекватное потребление кальция. При низком уровне кальция в моче диета должна быть обогащена кальцием и витамином D. Кроме того, хранение глюкоцереброзида в тканевых макрофагах может изменить образование компетентных остеокластов и привести к неспособности поддерживать здоровый костный матрикс. Прежде чем будет разработана полностью эффективная терапия костных осложнений, необходимы дальнейшие исследования для определения патогенеза этого заболевания. Редким явлением при заболевании типа 1 является внекостное распространение клеток Гоше на окружающие мягкие ткани, состояние, которое может имитировать злокачественный процесс (102).Поскольку частота нескольких злокачественных новообразований, включая лимфопролиферативные нарушения, увеличивается при болезни Гоше (114), для точного дифференциального диагноза может потребоваться биопсия.
Гипотеза о том, что костные кризы являются результатом прогрессирующего сосудистого нарушения, вызванного непосредственно закупоркой сосудов клетками Гоше, не подтверждается сцинтиграфическими или гистологическими исследованиями. Фактически, сканирование перфузии костей часто улучшается. Окклюзия сосудов клетками Гоше не объясняет комбинацию остеопении, остеонекроза и остеосклероза, наблюдаемую при заболевании.Другие сосудистые осложнения, такие как преждевременный инсульт, инфаркт миокарда или почечная недостаточность, не являются признаками заболевания, что делает простой процесс окклюзии сосудов менее привлекательным объяснением поражения скелета. Метаболические и эндокринологические исследования указывают на дисбаланс в гомеостазе кальция; однако не весь скелет поражается равномерно. Напротив, поражения состоят из скоплений клеток Гоше, разбросанных по костному веществу. Костные осложнения, вероятно, являются результатом токсического процесса вокруг этих очагов, который затем вторично ведет к отеку, сосудистой недостаточности и инфаркту.В одном исследовании сообщалось о корреляции между тяжестью заболевания типа 1 и уровнями цитокинов, полученных из макрофагов, в сыворотке крови. Эта корреляция предполагает, что дисбаланс цитокинов может играть роль в патофизиологии поражений костей (60). Исходя из имеющейся информации, маловероятно, что инфаркт вызван окклюзией сосудов клетками Гоше, а скорее сосудистым нарушением, указанным выше.
Прогрессирующий кифоз (иногда со сколиозом) без каких-либо аномалий тела позвонка обычно наблюдается у пациентов с типом Гоше 3.Это очень редко наблюдается у пациентов с типом Гоше 1. Требуется коррекция и стабилизация кифоза, и стержни Харрингтона часто помещают ближе к концу периода роста (поздний подростковый возраст). Механизм этого явления неизвестен, но исследования на модели рыбок данио предполагают аномалию развития костей (146).
Задержка роста и задержка созревания скелета наблюдаются у детей, но нормализуются с помощью заместительной ферментной терапии (69).
Органические системы прочие. В почках клетки Гоше можно найти в коре, мозговом веществе или клубочках. Почечная функция у пациентов с Гоше обычно нормальная. В некоторых случаях сообщалось о протеинурии или гематурии. Причина этих признаков неясна, но их связывают с инфильтрацией клеток Гоше.
Очаговые скопления клеток Гоше обнаруживаются внутри пейеровских бляшек и собственной пластинки желудочно-кишечного тракта. Пациенты могут жаловаться на вздутие живота, судороги и диарею. Аномальное усвоение кальция может быть одним из нескольких факторов, влияющих на костную ткань при заболевании.
Вишнево-красное пятно, которое появляется в макуле при некоторых сфинголипидозах и муколипидозах, не встречается при болезни Гоше; однако белые пятна, содержащие клетки Гоше, были замечены в стекловидном теле и сетчатке некоторых пациентов Гоше типа 3, и они могут развиваться, пока пациент находится на заместительной ферментной терапии (109). Сообщалось о случае, когда потерю зрения из-за увеита при заболевании типа 1 можно было обратить вспять с помощью заместительной ферментной терапии (17). Было много сообщений, предполагающих, что пациенты с болезнью Гоше имеют повышенный риск развития злокачественных новообразований, особенно опухолей кроветворения.Большое исследование 1525 пациентов выявило 2-3-кратный риск неходжкинской лимфомы, злокачественной меланомы и рака поджелудочной железы у пациентов с болезнью Гоше, но не обнаружил значимой связи между болезнью Гоше и раком в целом или с другими специфическими злокачественными новообразованиями, такими как множественная миелома (77).
Из-за различий в клиническом течении пациентов с болезнью Гоше исследователи попытались разработать лабораторный тест, который одновременно распознавал бы подтипы и был полезен для определения прогноза.Анализ генотипа дает некоторые рекомендации в этой области, но не является полностью удовлетворительным. У большинства людей даже с одним аллелем p.Asn409Ser неврологические нарушения отсутствуют, тогда как почти все пациенты с аллелем p.Leu483Pro имеют неврологические нарушения. Различия в эпидермальных клетках могут служить средством дифференциации нейронопатической болезни Гоше 2 типа от болезни Гоше 1 и 3 (46; 115). Дезорганизованные пластинчатые мембраны в роговом слое эпидермиса присутствуют у пациентов с типом 2, но не у пациентов с типом 3 (62; 24).
Гарви и его коллеги наблюдали, что амплитуда вызванных растяжением соматосенсорных вызванных потенциалов у пациентов с болезнью Гоше 3 типа положительно коррелирует со степенью когнитивного дефицита и, следовательно, с бременем неврологических заболеваний (50). Возможно, что измерение соматосенсорных вызванных потенциалов может быть полезным для мониторинга реакции будущих терапий, направленных на коррекцию невропатологии болезни Гоше.
Клиническая виньетка
Корпус 1. Эшли, 5-летняя девочка, перенесла инфекцию мочевого пузыря, которая стала очевидной, когда у нее внезапно начались несчастные случаи в туалете. Инфекция лечилась сульфамидными препаратами и сначала исчезла, но через 6 месяцев возникла повторно. Когда терапевт осмотрел Эшли, она обнаружила спленомегалию и низкое количество тромбоцитов. У Эшли диагностировали заболевание тромбоцитов, и ее лечили преднизоном. Через несколько дней после начала лечения стероидами количество тромбоцитов было очень низким, и была проведена биопсия костного мозга.Биопсия показала клетки Гоше. Последующее тестирование показало ферментный анализ 2 (NR 12-17) и генотип p.Asn409Ser / p.Leu483Pro.
Продолжение и обсуждение . Мутация p.Asn409Ser связана только с болезнью Гоше 1 типа. Когда p.Asn409Ser возникает со второй другой мутацией, такой как p.Leu483Pro, болезнь иногда проявляется в более раннем возрасте. У Эшли было 2 брата, и, поскольку ни у одного из них не было симптомов болезни Гоше, они не прошли генетическое тестирование. Когда генетическое заболевание, такое как болезнь Гоше, известно в семье, братья и сестры человека с этим заболеванием подвергаются более высокому риску иметь такое же заболевание и быть носителями.По этой причине для родителей важно рассказать всем детям в семье об этом заболевании. Многие родители хотят и нуждаются в помощи в этом процессе. Генетическое консультирование рекомендуется каждому члену семьи, когда он или она станет взрослым (или раньше, если есть показания).
Случай 2. В возрасте 4 месяцев родители Джастина отметили «что-то не совсем правильное с его движением глаз» и упомянули об этом своему педиатру во время 6-месячного визита Джастина к ребенку.
Во время этого визита педиатр заметил спленомегалию и услышал сообщение от родителей, что у Джастина был эпизод удушья, во время которого он стал цианотичным и был доставлен в отделение неотложной помощи.В своем развитии Джастин достиг ожидаемых этапов развития и в 6 месяцев уже сидел, хватался за предметы и откликался на свое имя. Педиатр назначил панель метаболических тестов, включая кислую бета-глюкоцереброзидазу.
Физикальное обследование | |||
• Печень примерно в 2 раза больше нормального размера | Лабораторные данные | ||
• Количество тромбоцитов 125 000 | |||
Последующие действия и обсуждение .Джастин лечился заместительной ферментной терапией в дозе 120 Ед / кг каждые 2 недели. Он хорошо себя чувствовал в течение нескольких месяцев; он шел хорошо и говорил несколько слов. В конце концов, он не смог прогрессировать, и у него стали чаще появляться приступы удушья. Джастин умер ночью в возрасте 22 месяцев.
Случай 3. Кристофер, мужчина европеоидной расы, 20 месяцев, поступил со следующим анамнезом: во время 12-месячного осмотра ребенка педиатр отметил гепатоспленомегалию.Гепатоспленомегалия Криса не изменилась при последующем наблюдении через 15 месяцев, а через 18 месяцев его осмотрели из-за раздражительности и лихорадки, которые сохранялись примерно 1 месяц. В то время были отмечены усиление гепатоспленомегалии и шейной лимфаденопатии. Кристофера поместили в больницу, где у него также была охриплость голоса и понос. Его обследовали на предмет возможного злокачественного новообразования и получили следующие результаты: рентген грудной клетки — нормальный; рентген брюшной полости, HSM; фрагментированный эпифиз правой бедренной кости; биопсия шейных лимфатических узлов в норме; аспират костного мозга, клетки Гоше.
Физикальное обследование | |
• 85 см, 10,5 кг | |
Продолжение и обсуждение . В течение следующих нескольких месяцев из обследования и наблюдений родителей стало очевидно, что у Кристофера был горизонтальный надъядерный паралич взгляда с компенсирующим толчком головы.Лечение заместительной ферментной терапией было начато в дозе 60 Ед / кг каждые 2 недели. Годы спустя, в возрасте 11 лет, у Кристофера не было прогрессирования медленных петлевых саккадических движений глаз, а также не было органегалии или явного заболевания костей; он играл в бейсбол и футбол, учился на отлично и лидировал в школьной игре в течение предыдущего учебного года.
Биологическая основа
Этиология и патогенез
Точные генетические и биохимические причины проявлений болезни и фенотипические различия между пациентами до конца не изучены.Нет никаких сомнений в том, что другие генетические модификаторы болезни Гоше вызывают заметные различия в представлении признаков и симптомов, связанных с костным мозгом, печенью, селезенкой, легкими, костями, мозгом и другими системами, вовлеченными в заболевание (53).
Ретикулоэндотелиальная система. Предполагается, что хранение гликолипидов в клетках ретикулоэндотелиальной ткани может вызывать воспалительную реакцию, характеризующуюся привлечением провоспалительных цитокинов и других клеток иммунной системы.В свою очередь, это может привести к обширному повреждению тканей, особенно печени, селезенки и костей. Исследование профиля экспрессии генов при болезни Гоше показало повышенную экспрессию генов, связанных с воспалительными реакциями в пораженной селезенке (91). В частности, значительно увеличилось количество транскриптов кДНК, представляющих цистеиновые протеиназы (катепсины B, K и S). Эти белки, как известно, участвуют в моделировании ткани, презентации антигена и, в случае катепсина К, в разрушении костного матрикса.
Вовлечение мозга. Понимание участия мозга в болезни Гоше за последние несколько лет расширилось, но оно далеко не полное из-за ограниченного числа посмертных случаев, доступных для изучения. Пациенты с болезнью Гоше 1 типа не имеют клинических симптомов или признаков, относящихся к нервной системе. В этой группе анатомические и биохимические исследования головного мозга проводятся нечасто. Глиоз гиппокампа был обнаружен в головном мозге неврологически нормальных пациентов с болезнью Гоше 1 типа (143).
При типах болезни Гоше 1, 2 и 3 периваскулярные клетки Гоше наблюдались в пространствах Вирхова-Робена (33; 143).
Патологические особенности нейронопатических фенотипов. В мозге пациентов с типом 2 были обнаружены свободные клетки Гоше в паренхиме, сопровождающиеся глиозом и микроглиальными узелками. Эти изменения присутствуют, но гораздо реже в мозге пациентов с типом 3. Накопление липидов в нейронах было предложено в нескольких отчетах, но это не было подтверждено ультраструктурно ни в одном случае болезни Гоше.При заболевании 2 типа сообщалось о нейронофагии и гибели нейрональных клеток в более глубоких слоях коры, таламуса, базальных ганглиев, ядер ствола мозга, мозжечка и спинного мозга. Различные степени демиелинизации описаны в головном мозге пациентов 2 типа. Из доступной информации можно было бы сделать вывод, что накопление кислой бета-глюкоцереброзидазы в головном мозге вызывает дисфункцию в окружающих клетках задолго до того, как наблюдаются дискретные патологические изменения (71). Обширное исследование 14 мозгов типа 1 (включая пациентов с паркинсонизмом), 2 и 3 болезни Гоше показало общие невропатологические находки при всех формах болезни Гоше (143).Были идентифицированы уникальные паттерны глиоза и потери нейронов, вовлекающие области CA2-4 гиппокампа и слой 4b калькариновой коры. Хотя эти данные были общими для всех трех фенотипов болезни Гоше, степень изменений варьировалась в зависимости от тяжести заболевания (143). У всех пациентов с болезнью Гоше были задействованы 3 и 5 слои коры головного мозга, CA2-4 гиппокампа и слой 4b. Потеря нейронов преобладала у пациентов как 2-го, так и 3-го типа с прогрессирующей миоклонической энцефалопатией, тогда как пациенты с болезнью Гоше 1-го типа имели только астроглиоз.Смежные области и пластинка, включая CA1 гиппокампа и калькариновую пластинку 4a и 4c, были избавлены от патологии, что подчеркивает специфичность уязвимости селективных нейронов. Повышенная экспрессия кислой бета-глюкоцереброзидазы с помощью иммуногистохимии была обнаружена в CA2-4. Была проведена авторадиография поглощения гиппокампа 45Ca (2+) в головном мозге крысы, которая продемонстрировала, что нейроны CA2-4 гиппокампа, а не нейроны CA1, были чувствительными к индуцированному кальцием высвобождению кальция (CICR-чувствительны). Эти результаты соответствуют биохимическим исследованиям, связывающим повышенные уровни глюкозилцерамида с сенсибилизацией рецепторов CA2-4 RyaR и 300% -ным потенциалом нейрональной чувствительности CICR (101).У 2 пациентов с болезнью Гоше 1 типа и паркинсонизмом в нейронах СА2-4 гиппокампа были обнаружены многочисленные синуклеин-положительные включения, подобные тельцам Леви стволового типа, обнаруженным при болезни Паркинсона. Эти данные свидетельствуют в пользу общего цитотоксического механизма, связывающего аберрантную кислотную бета-глюкоцереброзидазу, нейрональную цитотоксичность и образование цитотоксических телец Леви при болезни Гоше (143). Прогрессирование невропатологии очень предсказуемо на мышиной модели, включая активацию микроглии и астроглиоз, которые пространственно и временно коррелировали с избирательной потерей нейронов (44).Глюкоцереброзид накапливается в одинаковой степени в большинстве областей мозга, и при достижении определенного порога накопления в восприимчивых областях мозга начинается воспаление и нейродегенерация (43). Та же группа обнаружила, что некроз, опосредованный киназами взаимодействующих с рецепторами белков (RIP), играет важную роль в индукции воспаления и гибели нейронов при нейронопатической болезни Гоше (132). Дефицит Ripk3 (мыши с двойным нокаутом) резко улучшил клиническое течение мышей с болезнью Гоше, увеличив выживаемость и координацию движений.Следовательно, Ripk3 является новой терапевтической мишенью для нейронопатической болезни Гоше (132). Используя мышиную модель для наиболее тяжелой формы нейронопатии Гоше, было показано, что измененная лизосомная локализация и нарушение цитоскелета предшествуют нейровоспалительным путям, аксональной дистрофии и потере нейронов, ранее характерным для нейрональных форм болезни Гоше (148).
Глюкозилсфингозин вовлечен в токсическое повреждение нервных клеток. Эта молекула является метаболическим предшественником синтеза кислой бета-глюкоцереброзидазы, которая, как было показано, накапливается в коре головного мозга и мозжечка у пациентов с болезнью Гоше 2 и 3 типа (92; 92; 93) и у мышей Гоше.Ханнун и Белл показали, что лизосфинголипиды, включая глюкозилсфингозин, ингибируют протеинкиназу С, митохондриальную цитохром с оксидазу и CTP: фосфохолинцитидилилтрансферазу, которая, как предполагается, препятствует передаче сигнала и дифференцировке клеток (56).
Уровень глюкозилсфингозина не повышен у пациентов с болезнью Гоше 1 типа; однако уровень заметно увеличивается у пациентов с типом 3 (в среднем в 36 раз) и типом 2 (в 225 раз).Самые высокие уровни этого метаболита были обнаружены в ткани мозга 2 плодов с водянкой плода (98). Однако уровни глюкозилсфингозина в головном мозге слишком низкие и существенно не повышаются по сравнению с исходным уровнем в мышиной модели болезни Гоше 2 типа, и поэтому глюкозилсфингозин вряд ли будет участвовать в патогенезе нейронопатической болезни Гоше (101; 43).
Биохимические отклонения. Уровни кислой фосфатазы, ангиотензинпревращающего фермента, лизосомальных гидролаз, лизоцима и иммуноглобулинов повышены в плазме пациентов с болезнью Гоше (127; 95; 80; 136; 117; 89; 63; 107).Хитотриозидаза является особенно чувствительным индикатором накопления макрофагов у пациентов с болезнью Гоше (61). Из-за поражения печени у пациентов может быть увеличенное время частичного тромбопластина, протромбина и кровотечения. Кроме того, было высказано предположение, что повышенное количество глюкоцереброзида в плазме может мешать каскаду свертывания крови (18). Кислая бета-глюкоцереброзидаза повышается в плазме и может достигать 10 раз. Хотя было показано, что уровень глюкоцереброзида в плазме повышается после спленэктомии в некоторых норботнских случаях, о других подтипах заболевания не сообщалось.
Биохимические нарушения при нейронопатической болезни Гоше. Пациенты с типом Гоше 2 и 3 имеют такие же аномалии крови, как описано выше. В попытке идентифицировать биомаркеры, специфичные для неврологического вовлечения в болезнь Гоше, был идентифицирован неметастатический гликопротеин B (GPNMB) (147). Была обнаружена корреляция между тяжестью неврологических симптомов и уровнями GPNMB. То же самое было обнаружено на мышиной модели болезни Гоше. Эти данные предполагают, что GPNMB можно использовать в качестве маркера для количественной оценки невропатологии у пациентов с болезнью Гоше и в качестве маркера эффективности лечения в будущих исследованиях лечения (147).
Гематологические и скелетные проявления болезни Гоше происходят из-за макрофагального расстройства, тогда как аномалии мозга в основном вызываются вовлечением нейронов и астроцитов.
Хранение вещества. Глюкоцереброзид представляет собой соединение церамида и глюкозы. Фрагмент глюкозы этерифицируется с C-1 церамида по бета-глюкозидной связи. Соединение аналогично по структуре и свойствам группе сахаросодержащих липидов, выделенных из мозга Thudichum (126).
Цереброзиды состоят из церамида, этерифицированного множеством различных заместителей в C-1. Этот углерод может участвовать в реакциях с фосфорилхолином с образованием сфингомиелинов, незамещенного моносахарида или олигосахарида с образованием нейтральных гликосфинголипидов или олигосахарида, содержащего от 1 до 4 молекул сиаловой кислоты, с образованием ганглиозидов. Общей единицей среди этих соединений является церамид. Церамид получают из длинноцепочечного основания сфингозина (D (+) — эритро-1,3-дигидрокси-2-амино-4-трансоктадецен или сфингозин C18).Этот липид соединен амидной связью у С-2 с длинноцепочечной жирной кислотой с образованием церамида. Длина цепи жирных кислот варьируется. Обычно нейтральные гликосфинголипиды и сфингомиелины содержат жирные кислоты C20 и C24, тогда как ганглиозиды содержат жирные кислоты C18. Именно от сфингозина группа нарушений катаболизма липидов получила свое название (т.е. сфинголипидозы), потому что из него происходят накапливающиеся липидные соединения.
Глюкоцереброзид находится в конце катаболического пути гликосфинголипидов.Высшие гликосфинголипиды и ганглиозиды постепенно разлагаются специфическими кислотными гидролазами, что приводит к образованию глюкоцереброзида, который обычно разлагается до церамида и глюкозы под действием кислой бета-глюкоцереброзидазы. Соединения, которые способствуют накоплению глюкоцереброзида в периферических органах, — это глобозид, глоботриоза и лактозилцерамид. Они возникают в результате разрушения мембран, основным источником которых являются лейкоциты (70).
Важно отметить, что глюкоцереброзид, обнаруженный в селезенке, печени, почках, плазме и эритроцитах, содержит жирные кислоты с длиной цепи приблизительно от C22 до C24 (45).Глюкоцереброзид в мозге пациентов с заболеванием 2 типа состоит в основном из C18 (стеариновая кислота) (121; 93). Этот вывод был подтвержден и распространен на случаи 3-го типа (92; 29). Эти данные были интерпретированы как означающие, что глюкоцереброзид, накапливающийся в головном мозге, происходит из ганглиозидов внутри самого мозга. Это согласуется с известным содержанием жирных кислот в ганглиозидах. Было высказано предположение, что часть глюкоцереброзида в некоторых случаях типа 3 может быть получена из источников за пределами центральной нервной системы.Если данные подтвердятся, они должны иметь важные последствия, особенно потому, что было высказано предположение, что уровни глюкоцереброзида в плазме и тканях повышаются после спленэктомии в норботтнианских случаях (93).
Динамика накопления сфинголипидов
(Предоставлено доктором Джоном Барранджером)
Исследования патогенеза нейропатической болезни Гоше показывают, что дефектный гомеостаз кальция является механизмом, ответственным за нейропатофизиологию при острой нейронопатической болезни Гоше (101).Вызванное агонистами высвобождение кальция через рецептор рианодина было значительно увеличено в микросомах мозга от острой нейронопатической формы болезни Гоше (тип 2) по сравнению с подострой (тип 3) и ненейронопатической (тип 1) формами и контролями и коррелировало с уровни накопления GlcCer (101). Точный механизм, с помощью которого глюкоцереброзид усиливает высвобождение кальция через рецептор рианодина, не известен, но эти данные предполагают использование определенных блокаторов кальциевых каналов при нейронопатической болезни Гоше.Измененная экспрессия и распределение катепсинов в головном мозге нейронопатической мыши Гоше предполагает, что воспаление играет роль в заболевании и, следовательно, может быть целью терапевтического вмешательства (131). Повышение уровня GBA2, нелизосомальной глюкоцереброзидазы, может играть роль в заболевании (23).
Широкий спектр клинической тяжести у пациентов с болезнью Гоше, особенно у пациентов с типом 3, часто объясняется наличием генетических модификаторов.Интересная попытка использовала животные модели для идентификации таких генов (73). Авторы идентифицируют ряд вариантов, которые изменяют продолжительность жизни пораженной мыши. Наиболее интересным оказался Grin2b, который кодирует субъединицу NR2B рецептора N-метил-D-аспартата. Фармакологическое блокирование рецептора N-метил-D-аспартата с помощью мемантина привело к значительному удлинению модели мышей Гоше (73). Усилия по выявлению генетических модификаторов у пациентов с типом Гоше 3 продолжаются.
Кислая бета-глюкоцереброзидаза. В дополнение к 5 ‘и 3’ нетранслируемым областям кДНК гена GBA содержит 1548 пар оснований, кодирующих кислотную бета-глюкоцереброзидазу человека. Молекулярная масса 59 716, рассчитанная из 536 аминокислот, выведенных из последовательности кДНК, хорошо согласуется с оценкой SDS-полиакриламида продукта трансляции мРНК плаценты человека in vitro (42; 41). Были идентифицированы пять потенциальных сайтов гликозилирования (Asp-X-Ser / Thr). Количественный анализ углеводов показывает, что только 4 гликозилированы (124).Это было подтверждено прямым анализом аминокислотной последовательности (Б. Мартин, неопубликованные данные). В дополнение к аминокислотной последовательности структурного белка рамка считывания кДНК кодирует еще 39 аминокислот выше аминоконца. Этот сигнальный полипептид содержит гидрофобное ядро, состоящее из Gly-Leu-Leu-Leu-Leu, и, кроме того, имеет глицин в сайте расщепления пептидазой. Эти особенности согласуются со свойствами сигнальных пептидов других перемещаемых белков.Кроме того, последовательность кДНК подтверждает присутствие сигнальной последовательности 2 кДа, идентифицированной исследованиями импульсного мечения. Согласно базе данных мутаций генов человека в Институте медицинской генетики в Кардиффе, существует 332 мутации гена GBA , описанные как причина болезни Гоше, включая 253 бессмысленные или бессмысленные мутации, а также сплайс-мутации, делеции и сложные генные перестройки.
Эпидемиология
Все типы болезни Гоше наследуются по аутосомно-рецессивному признаку.Коллективные подтипы болезни Гоше составляют одну из наиболее распространенных форм сфинголипидозов и наиболее распространенное лизосомное нарушение накопления. Болезнь Гоше 1 типа, ненейронопатическая форма, встречается с частотой примерно от 1 из 40 000 до 60 000 среди населения в целом и от 1 из 500 до 1000 среди евреев-ашкенази. Пациенты с болезнью Гоше 3 типа (хроническая нейронопатическая болезнь Гоше) составляют около 5% популяции пациентов Гоше в Соединенных Штатах и в Европе, с оценочной заболеваемостью примерно 1 из 100 000.Однако сообщения предполагают, что нейронопатическая форма болезни Гоше преобладает в таких странах, как Китай, Япония, Корея, Тайвань, Египет и Сирия (134; 28; 123; 67; 36; 04; 55). Причина в том, что мутация N370S распространена в европейских странах, но почти отсутствует в Северной Африке и Азии, тогда как мутация L444P особенно распространена на Востоке (134; 28; 123; 67; 36; 04; 55).
Аутосомно-рецессивный тип наследования при болезни Гоше
(предоставлено Др.Джон Барранджер.)
Профилактика
Хотя болезнь Гоше 1 типа чаще встречается среди евреев Восточной Европы, в этой группе она обычно менее тяжелая. У чернокожих он тяжелее. Это обобщение полезно при консультировании, но есть исключения. Поэтому важно знать виды осложнений заболевания, которые произошли в семье. В целом, степень тяжести расстройства у братьев и сестер бывает одинаковой. Сообщалось об одной исключительной семье, в которой один случай типа 1 и один случай типа 2 произошли у полных братьев и сестер.
Значение для генетического консультирования. Рекомендуется тестирование и генетическое консультирование ближайших родственников больных. Следует указать на различия клинических подтипов и вариабельность некоторых форм заболевания. Из-за высокой несущей частоты среди ашкенази было предложено широкомасштабное тестирование несущей. Поскольку заболевание среди этой группы не всегда является катастрофическим (на самом деле, во многих случаях медицинская помощь оказывается во многих случаях только в позднем возрасте), этот вид тестирования, при желании, следует проводить в рамках системы, обеспечивающей тщательное консультирование, включающее адекватную информацию о беспорядок и объяснение ограничений обнаружения носителя.В гене GBA1 описано более 300 мутаций, 4 составляют большую часть нуклеотидных изменений. В еврейском населении ашкенази мутантный аллель p.Asn409Ser составляет около 75% аномальных генов (55). В этой этнической группе 4 аллеля (p.Asn409Ser [ранее описывался как Asn370Ser], c.84insG [ранее описывался как 84GG], p.Leu483Pro [ранее описывался как Leu444Pro] и IVS2 + 1G> A) составляют более 95 % хромосом, что позволяет проводить популяционный скрининг (11).В общей популяции обнаруживается много «частных» аллелей или мутаций, ограниченных одним родом. Тестирование на 4 перечисленные выше мутации приводит к выявлению менее 50% случаев. Таким образом, за исключением группы восточноевропейских евреев, скрининг населения не подходит для этого заболевания с использованием доступных в настоящее время технологий.
Пренатальная диагностика. Пораженные плоды любой формой болезни Гоше могут быть диагностированы пренатально с помощью ферментативного анализа культивированных амниоцитов или ворсинок хориона.Это особенно важно для семей, в которых наблюдались нейронопатические подтипы. Из-за сложности анализа носителей (32) и отсутствия соответствующих контрольных данных определение пренатального носителя с помощью ферментного анализа ненадежно. Однако молекулярно-генетические методы позволяют без труда эту идентификацию в семьях, в которых известны мутации.
Дифференциальный диагноз
Безболезненная спленомегалия является наиболее частым начальным признаком болезни Гоше 1 типа.Лимфопролиферативные злокачественные новообразования являются главной проблемой при диагностической оценке пациентов, поступающих таким образом. Результаты биопсии костного мозга могут указывать на присутствие клеток Гоше; однако диагноз должен быть установлен с помощью ферментативного анализа кислой бета-глюкоцереброзидазы в лейкоцитах или культурах фибробластов. Если пациент еврей, следует рассмотреть вопрос о болезни Гоше и провести ферментный анализ. Результаты обычно можно получить в течение нескольких дней. Заболевание, практически идентичное заболеванию Гоше, редко может быть вызвано дефицитом сапозина с, белка, активирующего глюкоцереброзидазу (129; 68).В этом случае активность глюкоцереброзидазы в норме, но у пациента есть клинические признаки, напоминающие болезнь Гоше. У младенцев с гепатоспленомегалией и нейродегенеративным течением болезнь Ниманна-Пика типа A и ганглиозидоз GM1 иногда похожи по клиническим проявлениям, но обычно их можно отличить по клиническим проявлениям.
Диагностическое обследование
Диагноз болезни Гоше следует рассматривать в любом случае необъяснимой спленомегалии с или без кровоточащего диатеза или других проявлений болезни скелета или печени.Расстройство следует рассматривать как вероятное у любого ребенка с гепатоспленомегалией и нейродегенеративным течением. Повышение уровня кислой фосфатазы, устойчивой к тартрату, в сыворотке крови свидетельствует о заболевании. Демонстрация характерных клеток Гоше в биоптатах костного мозга сужает диагностические возможности. Окончательный диагноз ставится с помощью анализа кислой бета-глюкоцереброзидазы в лейкоцитах, фибробластах, ворсинах хориона или моче.
Ячейка Гоше
Обратите внимание на смещение ядра к периферии и набухание клетки.(Предоставлено доктором Джоном Барранджером.)
После установления диагноза болезни Гоше необходимо оценить бремя болезни. Оценка характерных признаков заболевания должна включать гематологические, висцеральные, скелетные параметры и параметры качества жизни, как это рекомендовано Регистром болезней Гоше.
Гематологическое обследование. Гемоглобин, количество тромбоцитов, кислая фосфатаза, резистентная к тартрату, ангиотензинпревращающий фермент и хитотриозидаза должны быть измерены при установлении диагноза; Контрольные измерения рекомендуются каждые 3 месяца для пациентов, получающих заместительную ферментную терапию, и не реже, чем каждые 12 месяцев для пациентов, которые предпочитают не получать заместительную ферментную терапию.
Висцеральные исследования. Объем селезенки и печени также следует оценивать во время постановки диагноза. Рекомендуемый метод — объемная компьютерная томография или МРТ. Эти тесты рекомендуются каждые 12 месяцев для пациентов, получающих заместительную ферментную терапию, и каждые 24 месяца для пациентов, достигших стабильного состояния.
Ферментативный диагноз. Разнообразие естественных и искусственных субстратов обеспечивает точный анализ гомозигот, но все они имеют одни и те же ограничения при использовании для обнаружения гетерозигот (141; 32).Лучшим субстратом, используемым для анализа, является 4-метилумбеллиферил-бета-D-глюкопиранозид. Модификации и улучшения в первоначальном описании анализа, включающего добавление таурохолата, сделали его полезным в качестве диагностического инструмента и эквивалентом анализов с использованием природного субстрата. Анализ прост в использовании, а субстрат широко доступен, что делает его предпочтительным методом диагностической лаборатории. Кондуритол B-эпоксид является специфическим ингибитором кислой бета-глюкоцереброзидазы млекопитающих и позволяет подтвердить недостаточность фермента в системах, где может действовать неспецифическая бета-глюкозидаза.Лейкоциты и фибробласты могут быть приготовлены и отправлены в лаборатории для анализа в виде целых клеток или экстрактов. Разработан высокопроизводительный метод скрининга новорожденных (96).
С момента внедрения анализа кислой бета-глюкоцереброзидазы было замечено, что значения гетерозигот перекрывают таковые у контрольных субъектов по активности кислой бета-глюкоцереброзидазы в лейкоцитах и субпопуляциях лейкоцитов. Эта тема была подробно рассмотрена. Было разработано множество методов, но все они имеют одну и ту же проблему при обнаружении гетерозигот.В исследованиях известных гетерозигот примерно 20% носителей попадают в нормальный диапазон. Фибробласты обладают более высокой удельной активностью кислой бета-глюкоцереброзидазы, чем лейкоциты; однако широкий диапазон активности контрольных клеток, который изменяется со временем в культуре и условиях, делает эти клетки более непригодными для обнаружения гетерозигот. У тех, кому известен генотип, обнаружение носителя молекулярными методами полностью надежно.
Оценка скелета. Компьютерная томография, радионуклидное сканирование и магнитно-резонансная томография были полезны для оценки степени аномалий костей.Рекомендуемый метод для рутинной оценки заболевания костей — МРТ всех бедер с взвешиванием по Т1 и Т2 (25).
Плотность кости также следует оценивать у пациентов с болезнью Гоше. Исследования DEXA полезны для определения и оценки риска патологических переломов. Этим пациентам можно выполнить денситометрию позвоночника, бедра, предплечья и всего тела. Эти исследования следует проводить на исходном уровне и каждые 12 месяцев у пациентов, получающих заместительную ферментную терапию.
Менеджмент
Особый случай болезни Гоше 2 типа. Эта тяжелая форма болезни Гоше иногда вызывает серьезные проблемы с диагностикой, лечением и этическими проблемами. Они хорошо рассмотрены в 1 обзоре (140).
Трансплантация клеток и органов. Трансплантация селезенки и почек практически не повлияла на течение болезни у пациентов с болезнью Гоше. О трансплантации костного мозга сообщалось у пациентов с болезнью Гоше 1 и 3 типа. Достигнуто быстрое разрешение дефицита ферментов в циркулирующих лейкоцитах, что свидетельствует об успешном приживлении.
Показания к трансплантации гемопоэтических клеток различаются в зависимости от подтипа болезни Гоше. При заболевании 1 типа аллогенная трансплантация костного мозга приводит к разрешению органегалии в течение от нескольких месяцев до 2 лет. Болезнь костей также улучшается с исчезновением клеток Гоше из костного мозга, регрессом повреждений костей, улучшением линейного роста и исчезновением боли в костях (122; 105; 59). Таким образом, трансплантация костного мозга может быть излечивающей от заболевания типа 1, даже если этот терапевтический вариант ограничен доступностью доноров и высокими краткосрочными рисками, которые включают высокий процент связанных с процедурой заболеваемости и смертности около 15%.При заболеваниях типа 2 и 3 трансплантация костного мозга не влияет на неврологическое заболевание, и ее следует избегать (130).
Заменитель ферментов. Окончательная разработка заместительной ферментной терапии стала результатом удачного пересечения научных дисциплин, которые позволили сделать ключевые выводы:
• Открытие рецепторов для гликопротеинов (лектинов) (08) |
Фрагменты маннозы в замещающем ферменте олигосахариды являются ключом к успеху терапия.Манноза, а не манноза-6-фосфат, связывается с рецептором маннозы на клетках Купфера. В гепатоцитах также есть рецепторы маннозы, хотя некоторые из них не являются эндоцитарными. Следовательно, кислая бета-глюкоцереброзидаза может проникать как в гепатоциты, так и в клетки Купфера, но доставка к клеткам Купфера намного лучше для (модифицированной) кислой бета-глюкоцереброзидазы с концевым маннозом.
Углеводная единица нативной кислой бета-глюкоцереброзидазы
GlcNAc, N-ацетилглюкозамин; NeuNAc, N-ацетилнейраминовая кислота (сиаловая кислота).(Предоставлено доктором Джоном Барранджером.)
Ранние исследования замены ферментов породили большие надежды и воодушевление, что болезнь Гоше можно лечить с помощью инфузий ферментов (20). Первоначальные обнадеживающие биохимические и клинические отчеты не подтвердились. Несмотря на то, что количество фермента в последующих испытаниях было значительно увеличено, никаких изменений в ответ на действие фермента невозможно было измерить. Эти испытания не увенчались успехом, поскольку они не смогли направить фермент в нужную клетку в достаточном количестве.
Был разработан метод улучшения этого аспекта проблемы (48). Используя ферментативную деградацию олигосахаридных боковых цепей кислой бета-глюкоцереброзидазы, чтобы обнажить остатки маннозы, молекула фермента была адаптирована для связывания с природными рецепторами маннозы на плазматических мембранах макрофагов. В исследованиях на животных моделях эта сконструированная кислая бета-глюкоцереброзидаза доставлялась к ретикулоэндотелиальным клеткам в количествах, в 10 раз превышающих количество немодифицированного нативного фермента (48).Функция боковых цепей олигосахаридов до конца не изучена, но они, вероятно, важны для поддержания третичной структуры белка во время его синтеза и транслокации в лизосому. Если полностью предотвратить добавление боковых цепей сахара, белок не свернут должным образом и не будет иметь ферментативной активности.
Клиническое исследование с участием одного пациента продемонстрировало терапевтическое действие этой кислой бета-глюкоцереброзидазы с концевыми группами маннозы (10). Официальное клиническое исследование подтвердило эти результаты (13; 12).Коммерческий препарат в настоящее время широко используется у пациентов и успешно устраняет висцеральные, гематологические и скелетные проявления болезни.
Ответ на лечение кислой бета-глюкоцереброзидазой и синдром отмены при болезни Гоше
Сплошные линии на оси x указывают время введения фермента. (Предоставлено доктором Джоном Барранджером.)
После нескольких клинических испытаний кислой бета-глюкоцереброзидазы (альглюцеразы), полученной из плаценты, для лечения болезни Гоше, в 1991 г. была одобрена альглюцераза (12).Было достигнуто производство того же белка с использованием технологии рекомбинантной ДНК, и полученный препарат, имиглюцераза, получил одобрение регулирующих органов в 1994 году.
Обзор печеночных, селезеночных и гематологических результатов у 175 пациентов был проведен Weinreb и коллегами (139). Авторы пришли к выводу, что после 12-24 месяцев заместительной ферментативной терапии можно ожидать следующих 4 ответов:
(1) У пациентов с гепатомегалией объем печени уменьшается примерно на 20%. |
Лечение болезни Гоше с помощью ERT
На фотографиях показан пациент до и после заместительной ферментной терапии. Обратите внимание на уменьшение гепатоспленомегалии. (Предоставлено доктором.Джон Барранджер.)
Скелетные осложнения болезни Гоше хорошо поддаются лечению в те же сроки, что и другие признаки и симптомы болезни. Боль в костях уменьшается, а частота костного инфаркта резко снижается почти до нуля у пациентов, получавших 60 Ед / кг рекомбинантной бета-глюкоцереброзидазы человека каждые 2 недели до развития значительной структурной аномалии. Рентгенографическая оценка скелетных осложнений, однако, остается неадекватным инструментом для измерения исходного уровня и улучшения состояния кости в ответ на терапию.
Замещение фермента оказывает такое же благотворное влияние на пациентов с болезнью Гоше 3 типа, как и на пациентов типа 1 (111; 06). Этим пациентам показано лечение в дозе 60 МЕ / кг массы тела каждые 2 недели и должно тщательно контролироваться клинически и с помощью лабораторных исследований, чтобы установить ответ на терапию (130). При запуске на ранней стадии заболевания ферментативная заместительная терапия нормализует рост, корректирует гематологические нарушения и предотвращает скелетные осложнения, которые возникают у пациентов с болезнью Гоше 3 типа.Нет сомнений в том, что эта терапия заметно улучшила течение болезни у этой группы пациентов, которые умирали в подростковом возрасте до появления заместительной ферментной терапии. Однако при заболевании типа 2 заместительная ферментная терапия может продлить течение болезни, но неврологическое ухудшение прогрессирует, и плохой прогноз болезни не изменится; поэтому в этой группе не следует начинать заместительную ферментную терапию (104).
Побочные эффекты заместительной ферментной терапии немногочисленны и редко являются причиной для прекращения терапии (90).Антитела к заместительной ферментной терапии были обнаружены примерно у 15% пациентов. Частота серьезных осложнений очень низкая.
Эффекты прекращения и отмены ферментной терапии были оценены у пациентов, которые полностью прекратили терапию. Эти исследования предполагают, что полное прекращение терапии неизбежно подвергнет пациента опасности из-за ухудшения гематологических, биохимических и скелетных параметров (133; 137). Однако сообщалось об успешном опыте отмены ферментов на короткие периоды времени (112).
Раннее начало заместительной ферментной терапии очень важно для детей, поскольку она позволяет предотвратить серьезные необратимые скелетные осложнения у детей. У детей наблюдается значительное увеличение минеральной плотности костной ткани и скорости роста (69; 07).
Мышиные модели болезни Гоше. Модель мыши с дефицитом бета-глюкоцереброзидазы трансгенной кислоты, полученная путем целенаправленного нокаута гена кислой бета-глюкоцереброзидазы, демонстрирует тяжелый фенотип с пренатальной или перинатальной смертью (128).Ряд других моделей мышей использовались ограниченно и не представляли подлинной модели нейронопатической формы заболевания (144). Были созданы две истинные модели острой нейронопатической формы болезни Гоше. Одна из моделей имела нормальную ферментативную активность в гемопоэтических клетках, но не имела ферментативной активности в нейрональных и астроцитарных клетках. В этой модели не было значительного смягчения нейронального заболевания, что подтверждает наблюдение пациентов о том, что трансплантация костного мозга неэффективна при нейронопатической болезни Гоше (39).
Другие терапевтические подходы. Успех заместительной ферментной терапии, вероятно, привел к снижению мотивации к разработке подхода генной терапии для лечения нееврологических аспектов болезни Гоше. Поскольку существует неудовлетворенная потребность обратить вспять или предотвратить первичные неврологические осложнения болезни Гоше, продолжаются исследования по разработке подходов, которые позволят доставить кислую бета-глюкоцереброзидазу или нормальную копию гена GBA1 через гематоэнцефалический барьер и в клетки мозга (119).
Перенос гена Ex-vivo при болезни Гоше
Изображение иллюстрирует подход к переносу гена ex vivo при болезни Гоше. (Предоставлено доктором Джоном Барранджером.)
Лишение субстрата. N-бутилдезоксиноджиримицин (OGT-918), ингибитор глюкоцереброзид-синтазы, инициирует гликосфинголипидный путь и катализирует образование глюкоцереброзида (31). Обоснование этой терапии состоит в том, чтобы уменьшить образование глюкоцереброзида до уровней, при которых остаточная кислотная бета-глюкоцереброзидазная активность пациентов была бы достаточной для катаболизма субстрата.У большинства пациентов наблюдалась диарея, наиболее частый побочный эффект, и 2 пациента выбыли из исследования из-за этой жалобы. У некоторых пациентов развилась периферическая невропатия. Эти результаты нельзя сравнивать с результатами, полученными при заместительной ферментной терапии, которая нормализует или сильно влияет на клинические параметры с незначительными побочными эффектами (09; 88).
Терапия ингибированием субстратного синтеза
Изображение иллюстрирует терапию ингибированием синтеза субстрата как подход к лизосомным болезням накопления.(Предоставлено доктором Джоном Барранджером.)
В рандомизированном контролируемом исследовании было обнаружено, что миглустат неэффективен для лечения болезни Гоше 3 типа (110).
Гораздо более специфичный ингибитор церамидгликозилтрансферазы, аналог церамида элиглустат, продемонстрировал заметную эффективность, аналогичную ферментной заместительной терапии при дозе 60 МЕ / кг / 2 недели (Kamath et al 2014; 30). Он не имеет побочного действия миглустата, но его метаболизм зависит от генотипа цитохрома P450 CYP2D6.Элиглустат был одобрен FDA в 2014 году (103). Тартрат элиглюстата является субстратом P-гликопротеина, что, возможно, объясняет его плохое распределение в головном мозге (113). Новые соединения с аналогичной функцией, но с хорошим проникновением в мозг, разрабатываются и проходят испытания на людях (84). Venglustat в настоящее время проходит испытания на взрослых с болезнью Гоше 3 типа (идентификатор ClinicalTrials.gov: NCT02843035).
Фармакологические шапероны. Предлагаемый подход к лечению болезни Гоше и других лизосомных болезней накопления основан на введении конкурентных ингибиторов активного центра, которые способствуют правильной укладке и транспортировке мутантного белка (и даже молекул дикого типа).Этот процесс приводит к повышению уровня мутантного фермента, который затем может функционировать во время периода вымывания лекарства (145; 135). Единственный фармакологический шаперон, который в настоящее время проходит клинические испытания, — это амброксол в высоких дозах (81; 72). Он показывает некоторые перспективы при миоклонических формах болезни Гоше 3 типа (72). Однако никаких контролируемых исследований, подтверждающих клиническую пользу амброксола, не проводилось.
Болезнь Гоше 2 типа. Ни один из конкретных терапевтических подходов, описанных выше, не показал эффективности у пациентов с наиболее тяжелой формой болезни Гоше.Общее ведение пациентов с болезнью Гоше 2 типа очень сложно и было рассмотрено (140).
Генная терапия. Генная терапия с использованием аденоассоциированного вирусного вектора, вводимого интратекально, находится на продвинутой стадии планирования.
Особые соображения
Беременность
Пациенты с болезнью Гоше могут забеременеть и родить нормальных здоровых младенцев. Следует соблюдать меры предосторожности для женщин, страдающих анемией или тромбоцитопенией во время беременности.Обильное кровотечение — частое осложнение после родов. Тератогенный потенциал кислой бета-глюкоцереброзидазы не оценивался, и пациенткам, планирующим беременность, следует сообщить об этом факте. Пруденс предполагает, что беременным женщинам нельзя назначать заместительную терапию ферментами в течение первого триместра. Однако беременные пациентки, получавшие как минимум 2 формы заместительной ферментной терапии, родили нормальных детей (37; 38).
Анестезия
Пациенты с анемией, тромбоцитопенией или поражением легких подвергаются повышенному риску осложнений, связанных с анестезией и хирургическим вмешательством.Особенно поучителен случай с пациентом, которому до операции на сердце не был поставлен диагноз (87).
Пациент с гемофилией — обзор
Клинические испытания генной терапии гемофилии
Приблизительно 100 пациентов с гемофилией прошли клиническую оценку стратегий переноса генов. 34 Первоначальное испытание переноса гена ex vivo для FIX было проведено на двух субъектах в Китае в 1990-х годах. Γ-ретровирусный вектор FIX использовали для трансдукции аутологичных фибробластов кожи перед повторной имплантацией.Не наблюдалось значительного увеличения уровней FIX. С тех пор один пациент получил внутривенное введение аденовирусного вектора FVIII; плазмидный вектор использовали в качестве второго подхода ex vivo, в котором аутологичные фибробласты имплантировали в сальник, а небольшую группу пациентов с гемофилией лечили внутривенным γ-ретровирусом FVIII. Помимо транзиторной тромбоцитопении, связанной с инфузией аденовирусного вектора, побочных эффектов не было, но ни один из этих подходов не увеличивал уровни FVIII или FIX более чем через несколько дней, и даже тогда уровни были в диапазоне от 1% до 4%.
Напротив, испытания переноса генов с помощью AAV продемонстрировали терапевтическую эффективность. К настоящему времени проведено три завершенных испытания AAV FIX, первое — на скелетных мышцах, а последние два — на печени. Оба исследования, направленных на печень, первое с использованием доставки AAV-2 в печеночную артерию и второе системное внутривенное введение AAV-8, привели к более длительной экспрессии FIX на терапевтически значимых уровнях (от 2% до 10%) в некоторые пациенты. Действительно, в самом последнем исследовании печени AAV-8 терапевтические уровни FIX теперь сохранялись более 3 лет. 35 В настоящее время проводится шесть продолжающихся испытаний генной терапии FIX, все с использованием векторов AAV и в двух случаях с использованием трансгена FIX с усилением функции, кодирующего вариант FIX Padua, который имеет примерно в семь раз более высокую специфическую активность по сравнению с FIX дикого типа. Недавнее исследование генной терапии AAV FVIII задокументировало стойкие уровни FVIII от 20% до 200% у шести пациентов.
Несмотря на эти достижения, проблемы сохраняются (таблица 135.11). Наиболее значительным был иммунный ответ реципиента против векторного капсида.В некоторых случаях потенциальные реципиенты генной терапии исключаются из участия из-за доказательств предшествующего воздействия AAV (нейтрализующие анти-AAV антитела), но даже у тех, у кого нет задокументированного предыдущего иммунитета против AAV, у нескольких пациентов развился отсроченный цитотоксический Т-клеточный ответ с временная гепатотоксичность и последующее снижение экспрессии трансгена. Этот иммунный ответ может быть смягчен временной иммуносупрессией, и недавние данные исследования AAV-8 позволяют предположить, что короткого курса преднизона может быть достаточно для минимизации гепатотоксичности у большинства пациентов.Следует отметить, что ни у одного из этих пациентов не было иммунного ответа против FIX.
Будущее генной терапии гемофилии многообещающее. После двух десятилетий отличного прогресса в доклинических исследованиях появилась реальная надежда на достижение клинического успеха, по крайней мере, для генной терапии FIX.