Лежена синдром — это… Что такое Лежена синдром?
врожденный комплекс пороков развития, обусловленный нарушением структуры одной из хромосом группы В. Описан в 1963 г. Леженом с соавторами.
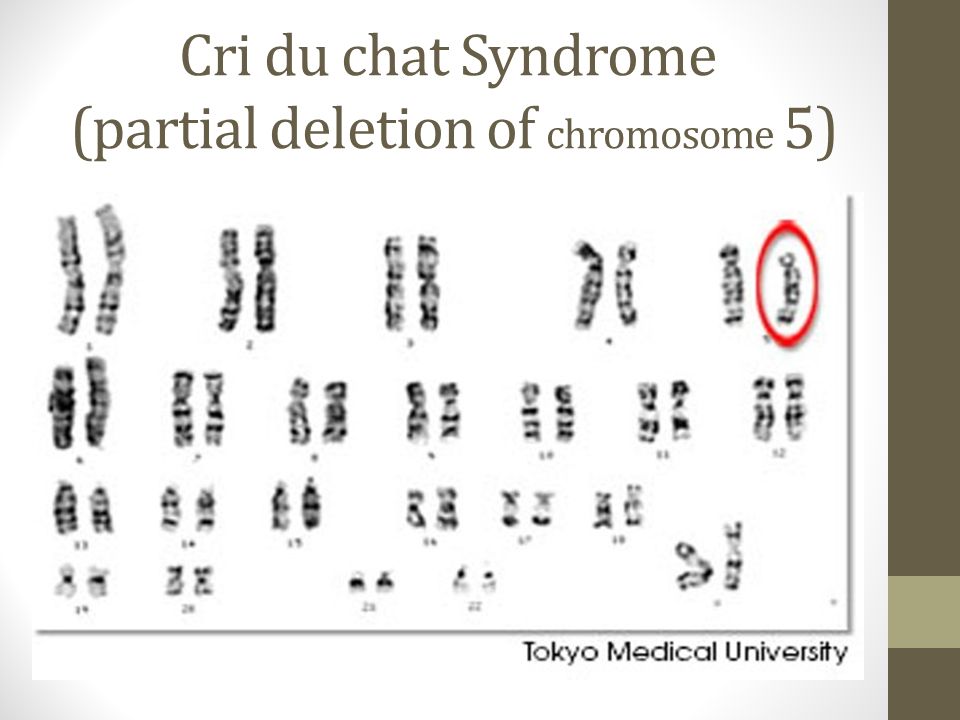
Частота синдрома среди новорожденных около 1: 3000, мальчики и девочки поражаются одинаково часто. Зависимость частоты рождения детей с Л.с. от возраста родителей не установлена. Синдром обусловлен изменениями короткого плеча хромосомы 5-й пары (рис. 1), возникающими чаще вследствие потери участка хромосомы, реже — структурной перестройки хромосомы или перемещения сегмента хромосомы внутри хромосомного набора. Описаны и другие варианты сбалансированных транслокаций в клетках родителей, приводившие к рождению детей с синдромом Лежена. Некоторая вариабельность клинических проявлений синдрома, по-видимому, зависит от размеров недостающего участка хромосомы.
Дети с Л.с. обычно рождаются с низкой массой тела (до 2500 г) даже при доношенной беременности. Наиболее постоянным признаком является специфический плач, напоминающий кошачье мяуканье.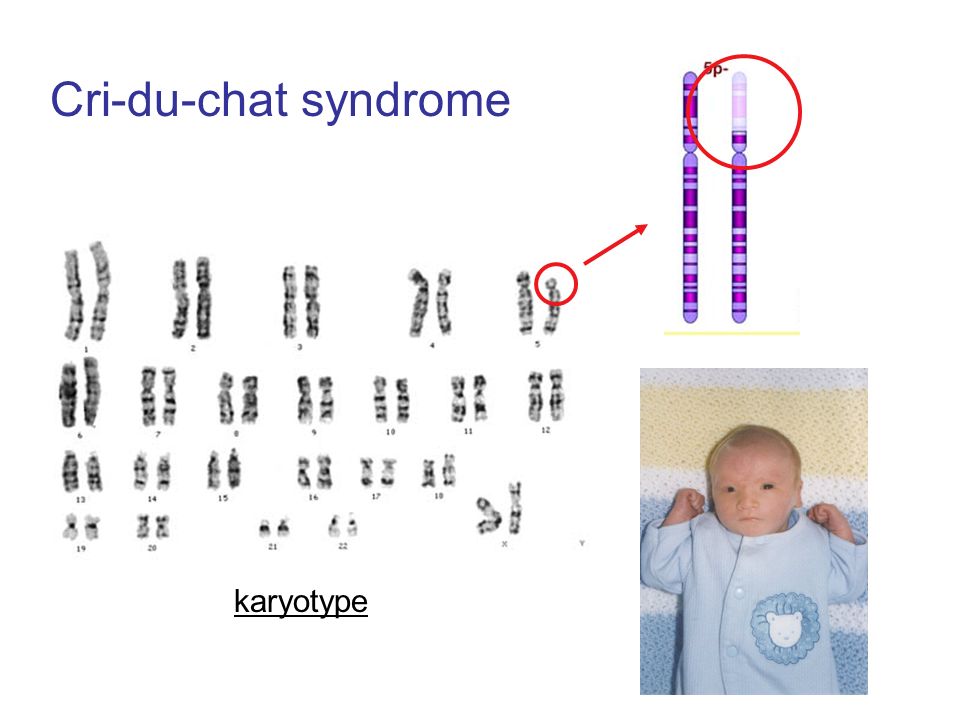 Этот симптом обусловлен особенностью строения гортани, определяемым при ларингоскопии — маленьким вялым надгортанником, который может опускаться над голосовой щелью. Голосовые складки не изменены. Рентгенологически отмечается уменьшение воздушного пространства над голосовыми складками.
Этот симптом обусловлен особенностью строения гортани, определяемым при ларингоскопии — маленьким вялым надгортанником, который может опускаться над голосовой щелью. Голосовые складки не изменены. Рентгенологически отмечается уменьшение воздушного пространства над голосовыми складками.
В раннем детском возрасте характерны лунообразное лицо, косой разрез глаз с опущенными наружными углами, эпикантус (складка у внутреннего угла глаза), гипертелоризм (широко расставленные глаза), несколько уплощенный нос, низко расположенные ушные раковины (рис. 2), впереди которых часто имеются небольшие (размером 1—3 мм) круглые фиброзные узелки. Мозговой череп относительно малых размеров (микроцефалия), долихоцефальной формы (значительное преобладание продольных размеров над поперечными) или с выступающими лобными буграми. Отмечается маленькая нижняя челюсть и короткая шея с избыточной кожей, формирующей крыловидные складки. В некоторых случаях отмечается расщепление верхней губы или неба либо высокое готическое небо и расщепление язычка.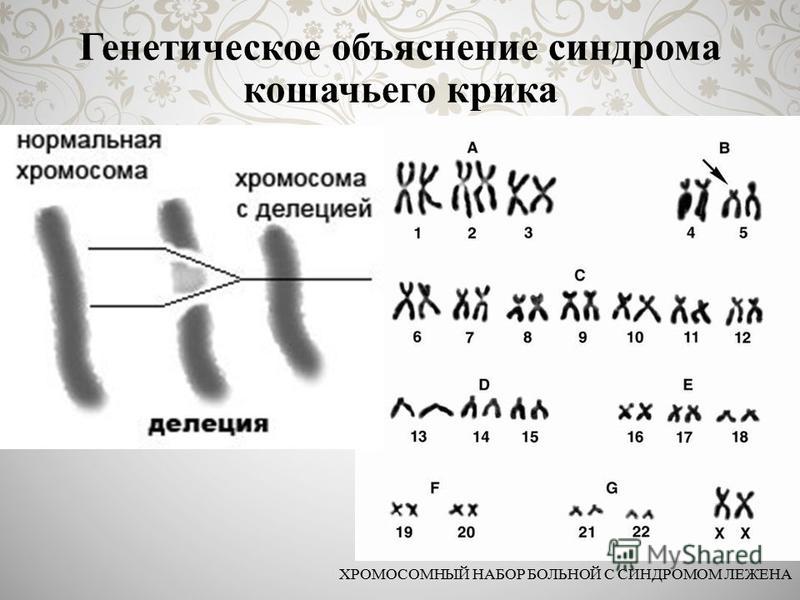 Возможны преходящее или постоянное Косоглазие, астигматизм (см. Рефракция глаза). В ряде случаев выявляют изменения глазного дна, в частности очаги депигментации сетчатки, а также атрофию зрительного нерва. Из аномалий развития внутренних органов наиболее часты пороки развития сердца и сосудов, почек. У мальчиков часто бывает Гипоспадия. Может быть четырехпалость или короткая, треугольной формы средняя фаланга V пальца. Общая мышечная гипотония, характерная для новорожденных с Л.с., обычно сохраняется в течение 1 года и дольше. У всех детей с Л.с. наблюдается умственная отсталость, большинство отстают в физическом развитии. Биохимические нарушения при Л.с. неспецифичны: длительное сохранение фетального гемоглобина, некоторое снижение содержания альбумина в сыворотке крови, умеренная аминоацидемия и аминоацидурия.
Возможны преходящее или постоянное Косоглазие, астигматизм (см. Рефракция глаза). В ряде случаев выявляют изменения глазного дна, в частности очаги депигментации сетчатки, а также атрофию зрительного нерва. Из аномалий развития внутренних органов наиболее часты пороки развития сердца и сосудов, почек. У мальчиков часто бывает Гипоспадия. Может быть четырехпалость или короткая, треугольной формы средняя фаланга V пальца. Общая мышечная гипотония, характерная для новорожденных с Л.с., обычно сохраняется в течение 1 года и дольше. У всех детей с Л.с. наблюдается умственная отсталость, большинство отстают в физическом развитии. Биохимические нарушения при Л.с. неспецифичны: длительное сохранение фетального гемоглобина, некоторое снижение содержания альбумина в сыворотке крови, умеренная аминоацидемия и аминоацидурия.
Частота и выраженность отдельных признаков Л.с. имеют возрастную зависимость. Такие признаки, как плач, напоминающий кошачье мяуканье, мышечная гипотония, лунообразное лицо в большинстве случаев с возрастом полностью исчезают, а микроцефалия, косой разрез глаз становятся более выраженными; прогрессирует отставание в психомоторном развитии.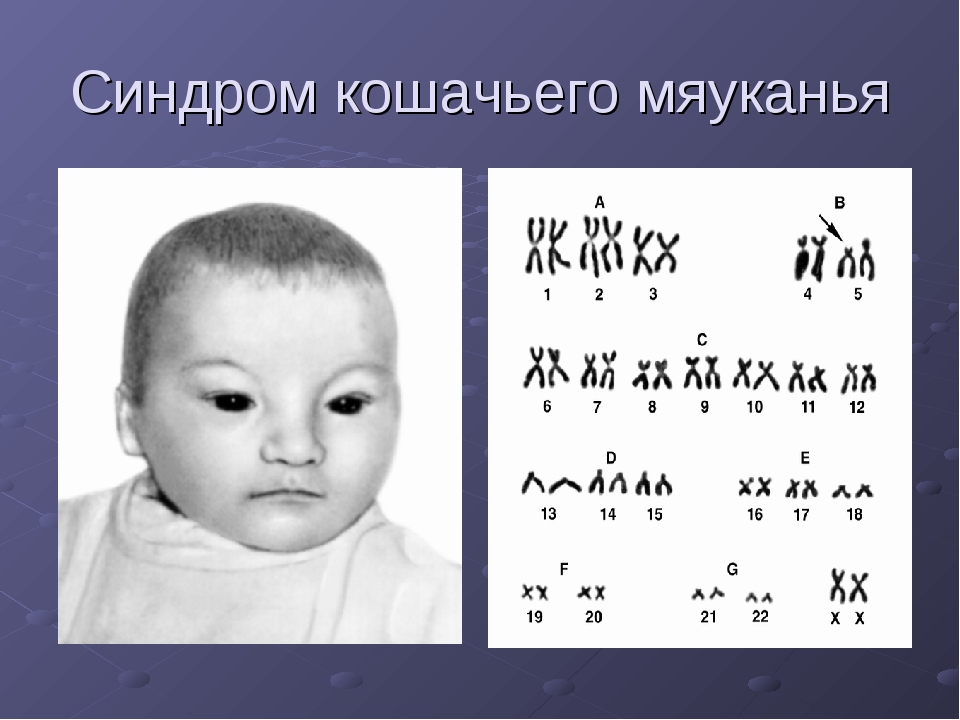 Может быть стридор; больные подвержены заболеваниям верхних дыхательных путей.
Может быть стридор; больные подвержены заболеваниям верхних дыхательных путей.
Синдром дифференцируют с другими врожденными пороками развития хромосомной и нехромосомной этиологии. Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом.
Лечение симптоматическое. Показаны средства, стимулирующие психомоторное развитие, лечебный массаж и гимнастика.
Профилактика заключается в своевременном проведении медико-генетического консультирования (Медико-генетическое консультирование) в семьях, где имелись больные с синдромом Лежена и основывается на определении кариотипа родителей, у которых был больной ребенок. Наличие изменений короткого плеча 5-й пары хромосом является абсолютным показанием для антенатального определения кариотипа плода при последующих беременностях путем амниоцентеза и исследования амниотических клеток. Сбалансированная транслокация у одного из родителей требует также исследования кариотипа у его кровных родственников с целью выявления лиц, имеющих транслокацию.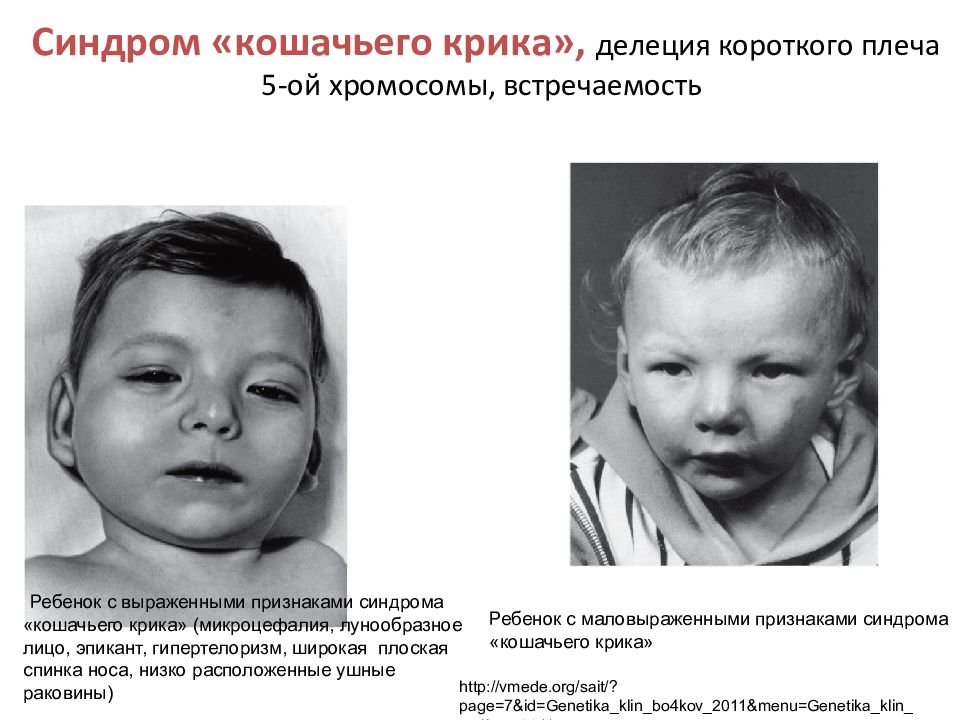
Библиогр.: Козлова С.И. и др. Наследственные синдромы и медико-генетическое консультирование, с. 337, М., 1987; Маринчева Г.С. и Гаврилов В.И. Умственная отсталость при наследственных болезнях, с. 180, М., 1988; Тератология человека, под ред. Г.И. Лазюка, с. 314, М., 1979.
набор больной с синдромом Лежена: групповая (от А до G) и индивидуальная идентификация хромосом (стрелкой указан дефект короткого плеча хромосомы 5-й пары, вторая хромосома не изменена)»>
Рис. 1. Хромосомный набор больной с синдромом Лежена: групповая (от А до G) и индивидуальная идентификация хромосом (стрелкой указан дефект короткого плеча хромосомы 5-й пары, вторая хромосома не изменена).
Рис. 2а). Ребенок с синдромом Лежена в возрасте 4 дней: лунообразное лицо, косой разрез глаз с опущенными наружными углами, несколько уплощенный нос, низко расположенные ушные раковины.
Рис. 2б). Ребенок с синдромом Лежена возрасте 4 лет.
4) Синдром кошачьего крика. Генетика Кариотип 46 XX или ху, 5р-.
 Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом.
Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом.
Хромосомно
синдром кошачьего крика объясняется
частичной моносомией; он развивается
при делеции (с утратой от трети до
половины, реже полная утрата) короткого
плеча пятой хромосомы. Для развития
клинической картины синдрома имеет
значение не величина утраченного
участка, а конкретный незначительный
фрагмент хромосомы. Изредка отмечается
мозаицизм по делеции или образование
кольцевой хромосомы-5.
Клиника
При этом синдроме наблюдается:
общее
отставание в развитии,
низкая
масса при рождении и мышечная гипотония,
лунообразное
лицо с широко расставленными глазами
характерный
плач ребёнка, напоминающий кошачье
мяуканье, причиной которого является
изменение гортани (сужение, мягкость
хрящей, уменьшение надгортанника,
необычная складчатость слизистой
оболочки) или недоразвитие гортани.
Признак исчезает к концу первого года
жизни. Кроме того, встречаются врожденные
пороки сердца, костно-мышечной системы
и внутренних органов, микроцефалия,
птоз, низкое расположение и деформация
ушных раковин, кожные складки впереди
уха, гипертелоризм (увеличенное расстояние
между какими-либо парными органами или
анатомическим образованиями (например,
между внутренними краями глазниц,
грудными сосками),
Билет 90
1)
В основе бесполого размножения организмов
лежит универсальный процесс – непрямое
деление клетки или митоз. Митоз
обеспечивает эквивалентное распределение
генетического наследственного материала
в ряду поколений клеток. Этот процесс
состоит из 2 главных этапов: митотического
деления ядра- кариокинеза и деления
цитоплазмы цитокинеза.
Фазы
митоза:
Профаза.
Происходит постепенная конденсация и
спирализация хромосом, постепенно
исчезает ядрышко. К полюсам клетки
расходятся дочерние центриоли, и между
ними формируется веретено деления.
Прометафаза.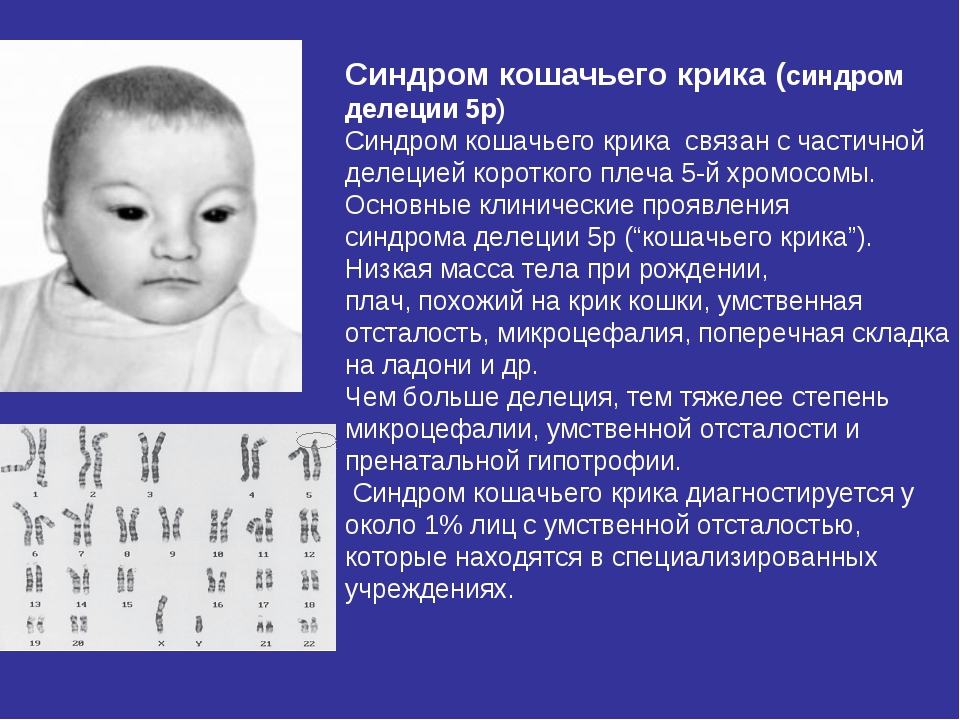
Ядерная о-ка распадается на несколько
фрагментов. В области центромер появляются
кинетохоры, функционирующие как центры
организации кинетохорных микротрубочек.
На этой стадии наблюдается передвижение
хромосом к экватору веретена.
Метафаза.
Хромосомы выстраиваются вплоскости
экватора клетки. Они хорошо заметны и
имеют вид толстых изогнутых палочек.
Анафаза.
В начале анафазы каждая хромосома
делится продольно на 2 хроматиды- дочерние
хромосомы, нити веретена деления начинают
сокращаться, а вместе с ними к полюсам
клетки начинают своё расхождение
хроматида каждой хромосомы. В поздней
анафазе около полюсов клетки начинают
собираться хромосомы, сохраняющие
расположение в виде звёзд.
Телофаза.
Хромосомы сближаются, образуя у полюсов
сначала форму рыхлого, затем плотного
клубка, появляются ядерные о-ки , хромосомы
деспирализируются, появляются ядрышки.
В цетре материнской клетки появляется
перегородка, растущая к боковым пов-тям
клетки.
2)Метод
гибридологического анализа:
Исходно:
брал чистые линии (гомозиготы –
одинаковые аллели генов)Оценивал
ограниченное кол-во признаковПрослеживал
наследование в ряду поколенийВел
строгий учет, статистическую обработку
данных
Первый
закон
– закон единообразия первого поколения
Чистые
линии с альтернативными признаками!
При
скрещивании двух гомозиготных организмов,
отличающихся по альтернативным вариантам
одного и того же признака, все потомство
от такого скрещивания окажется
единообразным и будет нести признак
одного из родителей.
Второй
закон –
закон расщепления
Скрещиваются
гибриды F1
При
скрещивании двух потомков первого
поколения между собой (двух гетерозиготных
особей) во втором поколении наблюдается
расщепление в определенном числовом
соотношении: по фенотипу 3:1, по генотипу
1:2:1
Третий
закон
– закон независимого наследования
При
скрещивании двух гомозиготных особей,
отличающихся друг от друга по двум и
более парам альтернативных признаков,
гены и соответствующие им признаки
наследуются независимо друг от друга
и комбинируются во всех возможных
сочетаниях
3)
Явление паразитизма, как и любой другой
экологический феномен, возникло разными
путями. С одной стороны, по-разному
развиваются взаимные адаптации паразитов
и хозяев в разных систематических
группах организмов — классах и типах,
с другой — различны направления эволюции,
ведущие к возникновению разнообразных
форм паразитизма.
Первый
подход к исследованию происхождения
паразитизма конкретен. Он рассматривается
Он рассматривается
при изложении материала по частной
паразитологии в разделах, посвященных
описанию характеристик типов и классов
паразитических организмов и их
экологических групп. Второй подход
вскрывает общие закономерности перехода
к паразитическому существованию вне
зависимости от систематического
положения организмов, занимающих новые
экологические ниши.
Наиболее
просто объясняется происхождение
эктопаразитизма.
Один из путей к этому — через увеличение
количества источников питания
с последующей их сменой. Так, многие
насекомые имеют колюще-сосущий ротовой
аппарат, питаясь соками растений. Но
питание за счет прокалывания ткани и
всасывания жидкости и есть способ
поглощения пищи всеми кровососущими
членистоногими, ряд которых, потребляя
кровь человека и теплокровных животных,
продолжает пользоваться также и соками
растений, s
Другой
путь, ведущий к эктопаразитизму, —
хищничество.
Активные хищники, осваивающие для
питания все более крупные жертвы,
становятся вначале временными, а затем
и постоянными эктопаразитами за счет
удлинения контактов с организмом
хозяина. Так, многие пиявки, ведущие
Так, многие пиявки, ведущие
себя как хищники по отношению к мелким
организмам, становятся паразитами более
крупных животных, питаясь их кровью.
Увеличение продолжительности питания
— основное направление перехода от
временного к постоянному эктопаразитизму.
Действительно, из большого количества
кровососущих форм членистоногих наиболее
длительное питание на хозяине характерно
именно для постоянных паразитов, степень
контакта которых с хозяевами наиболее
высока.
Иной
путь возникновения эктопаразитизма —
через усиление контакта так называемых
гнездовых
паразитов
с поверхностью тела хозяина. Животные,
обитающие в убежище другого вида, могут
питаться его перьями, волосами и
отпадающими чешуйками кожного эпидермиса.
Переход к постоянному обитанию на
поверхности тела хозяина дает паразиту
большие преимущества. Возможно, так
возник паразитизм пухоедов, власоедов
птиц и млекопитающих и группы клещей —
обитателей эпидермиса животных и
человека.
Основная
масса случаев эндопаразитизма в полостных
органах, имеющих связь с внешней средой,
представляет собой явление, развившееся
в результате случайного
заноса
в организм цист, яиц или личинок
свободноживущих видов, предварительно
имеющих адаптации к обитанию в почве
или в воде, содержащей избыток органического
вещества.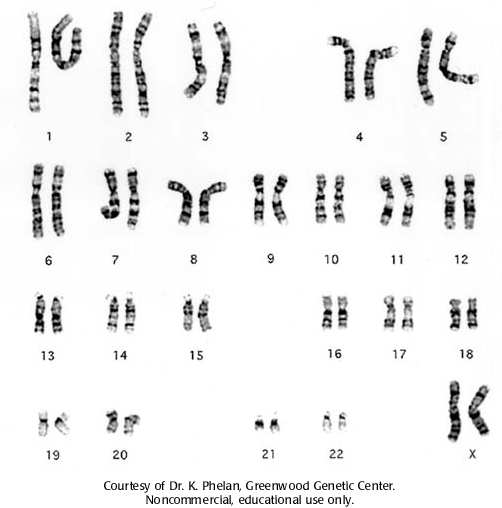 Примером является угрица
Примером является угрица
кишечная, которая в своем развитии
сохранила возможность обитать и
размножаться как в почве, так и в организме
человека
Возможен
переход к паразитированию в одном
хозяине после предварительной
адаптации к обитанию в другом,
служащем источником питания первого.
Так, известен целый ряд гельминтов,
которые, обитая в кишечнике рыбы, не
перевариваются в пищеварительной
системе хищников, съевших паразитов
вместе с хозяином и продолжающих
паразитировать в кишечнике или тканях
уже нового вида
Не
исключается и вариант перехода к
полостному паразитизму видов,
предварительно
адаптированных к эктопаразитизму.
Этим путем, вероятно, эволюционируют
некоторые насекомые, большую часть
цикла развития проводящие в ротовой
полости птиц, но выходящие для размножения
на перьевой покров их головы.
Наиболее
сложно и многообразно происхождение
паразитов тканей
внутренней среды.
Один из путей — через изменение инстинкта
откладки яиц и предварительных адаптации
к эктопаразитизму. Таким путем, вероятно,
Таким путем, вероятно,
произошел тканевой паразитизм личинок
мух и оводов, откладывающих яйца на
поверхности кожи и слизистых оболочек
животных и человека. Личинки при этом
вскоре погружаются под покровы и ведут
типичный эндопаразитический образ
жизни.
Многие
паразиты приспособились к обитанию в
тканях после освоения полостных органов,
связанных с внешней средой. Так,
по-видимому, шла эволюция паразитизма
у ленточных червей, у трихинеллы
спиральной. В цикле развития этих
паразитов имеются формы, обитающие как
в кишечнике, так и в тканях.
Некоторые
паразиты внутренней среды возникли,
вероятно, предварительно адаптировавшись
к обитанию в пищеварительной системе
членистоногих, а с переходом последних
к гематофагии заселили новую и
труднодоступную экологическую нишу —
кровь и другие ткани мезодермального
происхождения.
Таким
образом, путей перехода к паразитизму
у разных видов животных много, но
несомненным остается одно: паразитизм
— явление вторичное.
Об этом свидетельствует наличие в
жизненных циклах многих, даже наиболее
специализированных паразитов,
свободноживущих стадий, рекапитулирующих
свободный образ жизни предков .
Синдром кошачьего крика
Синдром кошачьего крика (англ. cri du chat syndrome, фр. maladie du cri du chat; также болезнь кошачьего крика, синдром Лежёна — по имени описавшего его в 1963 году французского учёного) — редкое генетическое расстройство, вызываемое отсутствием фрагмента 5-й хромосомы.
Эпидемиология
Частота проявления синдрома примерно 1 : 45 000. Соотношение полов М1 : Ж1,3.
Генетика
Кариотип 46 XX или XY, 5р-. Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом.
Хромосомно синдром кошачьего крика объясняется частичной моносомией; он развивается при делеции (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а утрата конкретного короткого фрагмента хромосомы. Изредка отмечается мозаицизм по делеции или образование кольцевой хромосомы-5.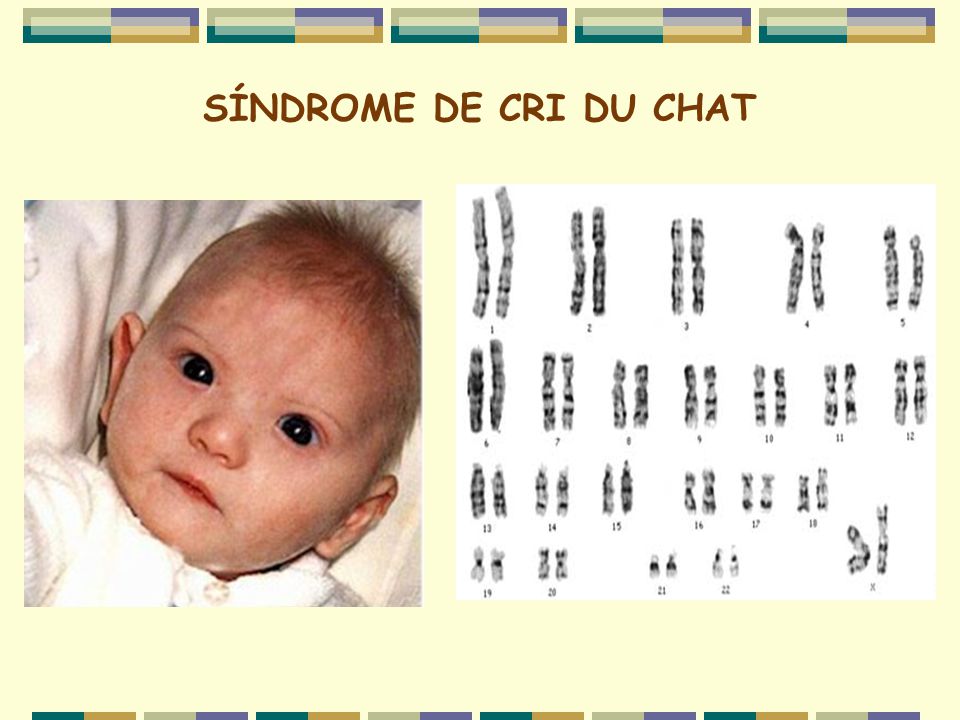
Клиника
При этом синдроме наблюдается:
- общее отставание в развитии,
- низкая масса при рождении и мышечная гипотония,
- лунообразное лицо с широко расставленными глазами,
- характерный плач ребёнка, напоминающий кошачье мяуканье, причиной которого является изменение гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани. Признак исчезает к концу первого года жизни.
Кроме того, встречаются врождённые пороки сердца, костно-мышечной системы и внутренних органов, микроцефалия, птоз, низкое расположение и деформация ушных раковин, кожные складки впереди уха, гипертелоризм (увеличенное расстояние между какими-либо парными органами или анатомическими образованиями — например, между внутренними краями глазниц, грудными сосками), эпикантус (вертикальная кожная складка около внутреннего угла глаза, обычно двусторонняя; наиболее чётко выражена при синдроме Дауна), антимонголоидный разрез глаз.
Клиническая картина синдрома и продолжительность жизни людей с этим синдромом довольно сильно варьируются по сочетанию врождённых пороков развития органов.
Лечение
Лечение симптоматическое. Показаны средства, стимулирующие психомоторное развитие, лечебный массаж и гимнастика.
Хромосомные нарушения
Хромосомные нарушения — это клинические синдромокомплексы, в основе которых лежат нарушения числа или структуры хромосом, то есть избыток или нехватка генетического материала, локализованного в той или иной хромосоме.
В норме у человека число хромосом равно 46, из которых 23 ребенок получает от матери и 23 аналогичные хромосомы от отца. В этом наборе гентического материала есть 2 особые хромосомы, которые были названы «половыми». Они определяют пол ребенка и ряд других важных признаков.
Таким образом, изменения числа хромосом (больше или меньше 46), а также изменение структуры хромосом (например, выпадение или удвоение даже небольшого кусочка хромосомы) получили название «хромосомные мутации».
Наиболее часто из них встречаются изменения модального числа хромосом — это отсутствие в хромосомном наборе какой-либо хромосомы (моносомия) или появление добавочной хромосомы (трисомия, тетрасомия и т.д.).
Число возможных изменений структуры хромосомы неисчислимое множество. К примеру, транслокации (обмен сегментами между разными хромосомами), делеции (выпадение участка хромосомы), дупликации (удвоение части хромосомы), инверсии (переворот сегмента хромосомы на 180 градусов) и т.д.
Хромосомные мутации, возникшие в половых клетках (сперматозоидах или яйцеклетках) или на первых этапах деления клеток зародыша, как правило, передаются большинству клеток развивающегося организма, вызывая множественные аномалии развития, а многие хромосомные изменения плода могут стать причиной спонтанных абортов и выкидышей, что важно учитывать в семьях, воспитывающих детей с задержками развития.
К факторам риска, способствующим их возникновению, относят ионизирующую радиацию, инфекции и интоксикации матери, эндокринные нарушения, психические травмы, воздействие ряда лекарственных препаратов и некоторых физиотерапевтических методов лечения.
Наиболее точно установлено, что причиной появления ребенка с хромосомными мутациями является не молодой возраст матерей (свыше 40 лет).
В последнее время очень большое значение придается факту скрытого носительства хромосомных нарушений у родителей родившегося ребенка (сбалансированные транслокации, мозаицизм). Изучение данного вопроса позволяет предотвратить риск повторного рождения ребенка с аналогичной формой заболевания.
Различают хромосомные синдромы, обусловленные изменением половых хромосом, и синдромы, вызванные аномалиями аутосом (любой из 44 неполовых хромосом).
Основными клиническими проявлениями аутосомных аномалий являются признаки психического и физического недоразвития, дисплазии (неправильное развитие), врожденные пороки развития (аномалии) и умственная отсталость различной степени тяжести. К врожденным порокам можно отнести: аномалии развития сердца, удвоение почки, расщелина неба, особенности строения кистей и стоп и многие другие. При заболеваниях, обусловленных нарушениями в системе половых хромосом, как правило, более характерны недоразвитие половых желез и аномалии развития вторичных половых признаков, также с симптомами задержки психо-речевого развития.
При заболеваниях, обусловленных нарушениями в системе половых хромосом, как правило, более характерны недоразвитие половых желез и аномалии развития вторичных половых признаков, также с симптомами задержки психо-речевого развития.
Различные хромосомные синдромы встречаются с разной частотой. По сводным данным многих исследований, распространенность наиболее частых из них среди новорожденных следующая:
трисомия по 21 хромосоме (синдром Дауна) 1:500
XXX (трисомия-Х) 1:1000 (девочек)
ХYY (синдром дубль-Y) 1:1000 (мальчиков)
ХХY (синдром Клайнфелтера) 1:1400 (мальчиков)
Х0 (синдром Шерешевского-Тернера) 1:3300 (девочек)
46,5р del (синдром «кошачьего крика») 1:4000
трисомия по 18 хромосоме (синдром Эдвардса) 1: 6800
трисомия по 13 хромосоме (синдром Патау) 1:7600
Не смотря на казалось бы не частую встречемость каждого отдельного синдрома, в целом хромосомные болезни у новорожденных наблюдаются не редко — с частотой около 1 : 100.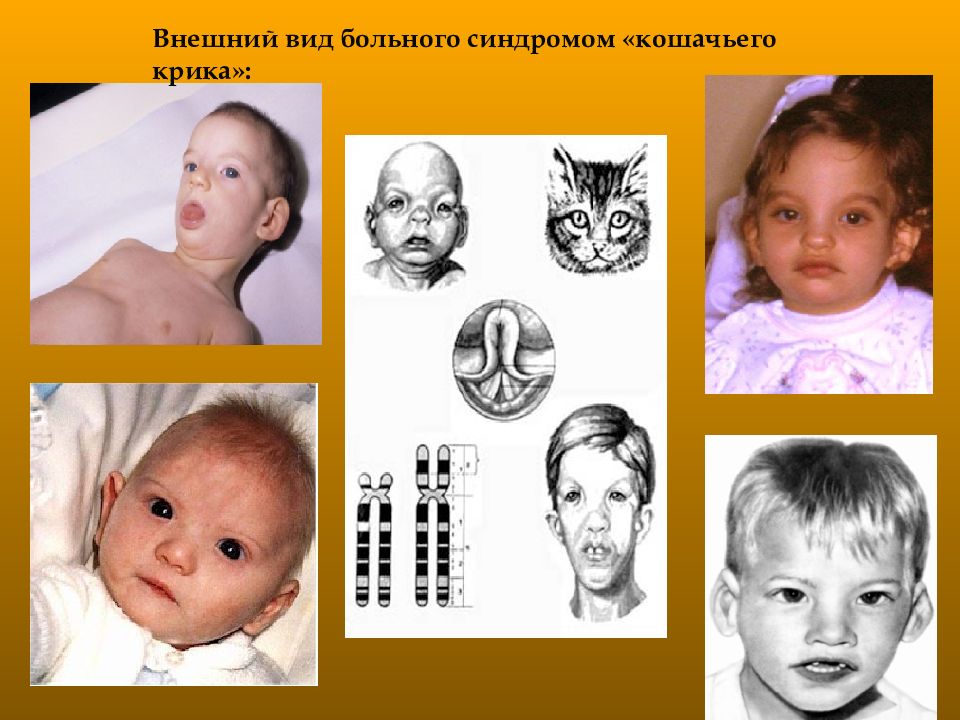 Ежегодно в России рождается свыше 30 тыс. детей с хромосомной патологией. Спонтанные выкидыши являются результатом хромосомной патологии в более чем 50%.
Ежегодно в России рождается свыше 30 тыс. детей с хромосомной патологией. Спонтанные выкидыши являются результатом хромосомной патологии в более чем 50%.
Рассмотрим основные клинические проявления отдельных хромосомных синдромов, сопровождающихся умственной отсталостью и задержками психо-речевого развития.
Синдром Дауна — врожденное заболевание, характеризующееся умственной отсталостью и рядом признаков эндокринной недостаточности.
Синдром впервые описан английским врачом Дауном в 1866 г. Встречается с частотой 1 на 500 новорожденных. Частота встречаемсоти у мальчиков и девочек одинакова. В основе заболевания лежит аномалия хромосомного набора (47 вместо 46). Лишняя хромосома обнаруживается в 21 паре, в связи с чем этот синдром иногда называют «трисомией по 21-й хромосоме» (47, 21+). Выявлена связь частоты рождения больных с увеличением возраста матери. Приблизительно в 3—4% случаев отмечаются транслокационные формы синдрома Дауна, при которых общее число хромосом в кариотипе нормальное — 46, а дополнительная 21-я хромосома транслоцирована (присоединена) на другую аутосому. Это является результатом того, что один из фенотипически здоровых родителей является скрытым носителем сбалансированной транслокации. Именно за счет этих форм повышается риск повторного рождения больного ребенка у молодых матерей. Еще 3-4% случаев синдрома Дауна составляют мозаичные варианты, при которых в организме одновременно обнаруживают и трисомные, и нормальные клетки. Порой, при небольшом проценте трисомных клеток ребенок с ЗПРР внешне может выглядеть абсолютно нормальным.
Это является результатом того, что один из фенотипически здоровых родителей является скрытым носителем сбалансированной транслокации. Именно за счет этих форм повышается риск повторного рождения больного ребенка у молодых матерей. Еще 3-4% случаев синдрома Дауна составляют мозаичные варианты, при которых в организме одновременно обнаруживают и трисомные, и нормальные клетки. Порой, при небольшом проценте трисомных клеток ребенок с ЗПРР внешне может выглядеть абсолютно нормальным.
Установлено, что для синдрома Дауна характерно уменьшение размеров и веса головного мозга, а также аномалии развития мозга и мозговых сосудов. Отмечаются также структурные изменения в железах внутренней секреции, печени и сердце. Клиническая картина синдрома Дауна характеризуется проявлениями симптомов умственной отсталости. Характерен также и внешний вид таких больных: косо расположенные глазные щели, широкая уплощенная переносица, дополнительная кожная складка у внутреннего угла глаз, высокое стояние твердого неба (признаки эмбриональной задержки в развитии лицевого скелета), полуоткрытый рот, увеличенный высунутый язык с выраженными сосочками и глубокими бороздами (признаки дисфункции щитовидной железы), выпадение волос (дисфункция надпочечников), низкий рост, короткая шея, укороченные кисти и стопы, искривление мизинца, на ладонях имеется поперечная складка, на стопах увеличен промежуток между 1 и 2 пальцами, выражены внешние проявления гипогенитализма.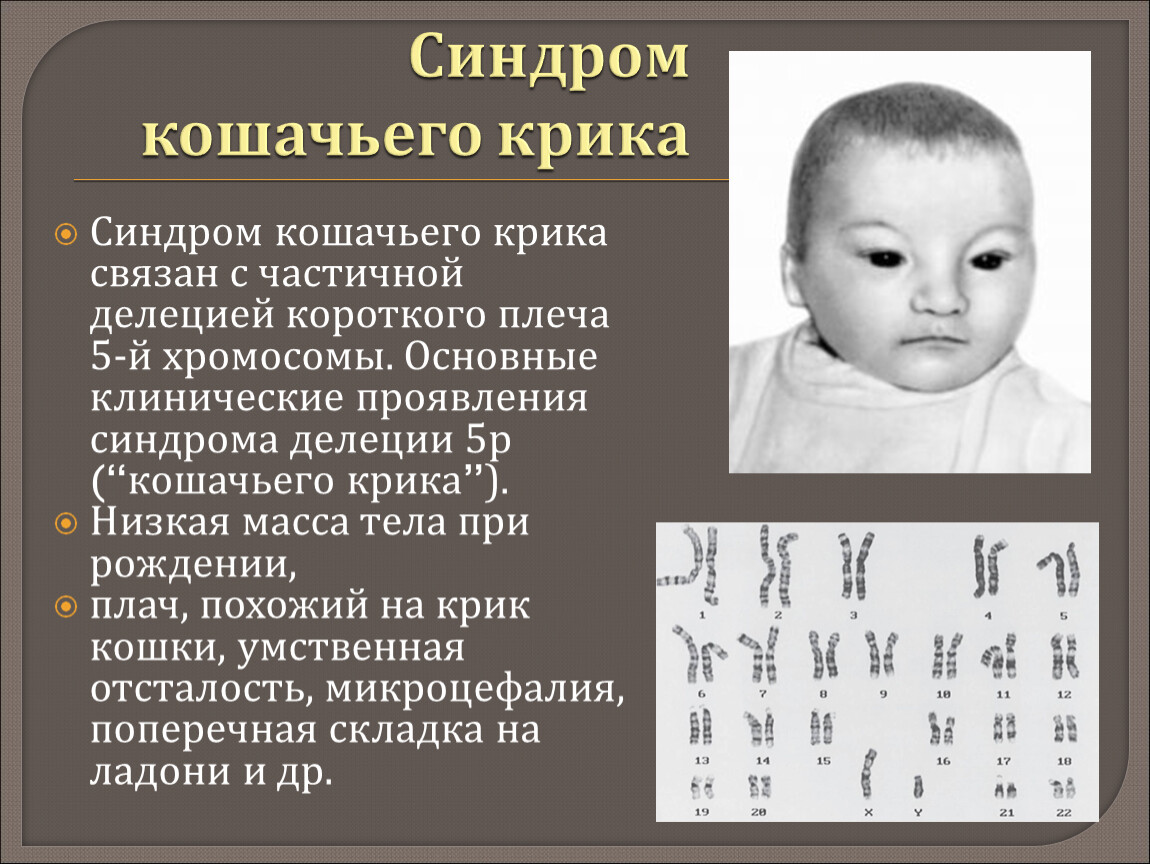
Такие дети с рождения отстают в росте, начинают поздно держать голову, сидеть и ходить. Речь, как правило, невнятная, словарный запас беден, произношение с дефектами в связи с недоразвитием высших мозговых функций, с одной стороны, и анатомическими аномалиями ротовой полости — с другой.
В клинической картине заболевания доминируют симптомы неврологической патологии, диффузная мышечная гипотония (снижение мышечного тонуса), благодаря чему больные гибки и иногда могут складываться как «перочинный ножик», расстройства координации движений, косоглазие, выраженные вегетососудистые нарушения.
Особенностью психического дефекта является относительная сохранность эмоциональной сферы по сравнению с тяжестью интеллектуального недоразвития. Так, больные ласковы, добродушны, послушны. Характерной особенностью таких детей является повышенная внушаемость, что является положительным фактором при проведении коррекционной работы и отрицательным при их развитии.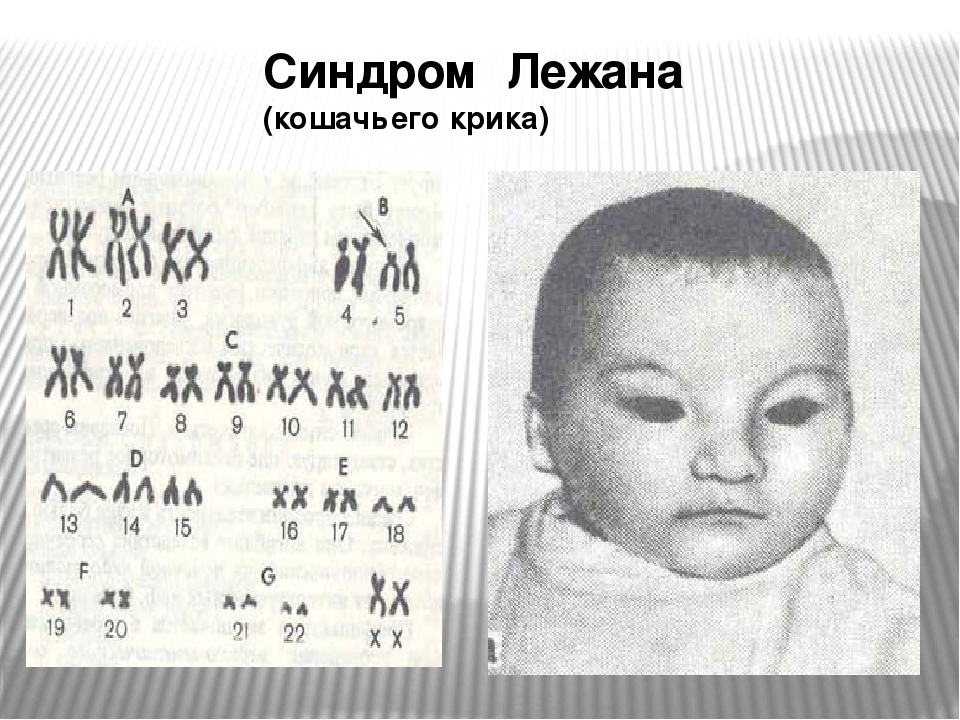
Уровень социального развития больных с синдромом Дауна зависит от степени и формы заболевания. Так, дети с более легкими формами умственной отсталости, хотя и медленно, но развиваютя, приобретая определенные навыки, знания, осваивая программу нескольких классов вспомогательной школы. Однако, как правило, большинство из них не достигают удовлетворительного уровня социальной адаптации и нуждаются в постоянной опеке. Им может быть оформлена инвалидность детства с момента точной диагностики заболевания. Особенностью возрастной динамики синдрома Дауна является позднее половое созревание и раннее появление признаков инволюции (25—30 лет). Мужчины с синдромом Дауна бесплодны, женщины могут давать потомство, половина которого также страдает синдромом Дауна.
Синдром Шерешевского—Тернера — симптомокомплекс проявлений врожденного, наследственно обусловленного недоразвития половых желез и передней доли гипофиза в сочетании с аномалиями соматического развития.
Впервые заболевание описано отечественным эндокринологом Н.А. Шерешевским (1925), а более подробно — американским эндокринологом Н. Тернером (N.H. Terner) л 1938 г. В основе заболевания лежит отсутствие одной хромосомы (половой Х-хромосомы) (45 вместо 46).
Клиническая картина синдрома характеризуется разной степенью умственной отсталости и ЗПРР, низким конечным ростом (135—145 см), замедлением полового развития, недоразвитием половых желез, аменореей, бесплодием и отсутствием грудных желез. Диспластические расстройства проявляются в виде короткой шеи и особых кожных складок, идущих от затылка к надплечью, укорочением 4 пальцев на руках и искривлением мизинцев, выраженной деформацией ушных раковин, наличием множественных пигментных родинок. Преимущественно данным синдромом страдают лица женского пола.
Синдром Клайнфелтера — заболевание, обусловленное нарушением числа половых хромосом (добавочные Х-хромосомы) (от 47 до 49), характеризующееся умственной отсталостью, нарушением смерматогенеза, недоразвитием яичек и вторичных половых признаков, а также нарушением пропорций тела. Впервые синдром описан американским эндокринологом Клайнфелтером (H.F. Klinfelter) в 1942 г. Его частота, по сводным данным, составляет до 2% среди умственно отсталых и до 0,5% (кадждый двухсотый мужчина) в среднем в мужской популяции.
Впервые синдром описан американским эндокринологом Клайнфелтером (H.F. Klinfelter) в 1942 г. Его частота, по сводным данным, составляет до 2% среди умственно отсталых и до 0,5% (кадждый двухсотый мужчина) в среднем в мужской популяции.
Клинические проявления синдрома Клайнфельтера варьируют от внешне нормального и интеллектуального развития до выраженного евнухоидизма и умеренной умственной отсталости. Однако в ряде случаев уже в раннем возрасте у больных отмечаются характерные своеобразные симптомы физического развития: низкий и узкий лоб, густые и жесткие волосы, высокое стояние таза, короткая, плоская и узкая грудная клетка, недоразвитие половых органов. Более отчетливо вышеперечисленные симптомы начинают обнаруживаться в подростковом, пубертатном возрасте. Характерен внешний вид взрослого больного с синдромом Клайнфельтера: высокий рост, астеническое сложение, узкие плечи, широкий таз, удлиненные конечности, слаборазвитая мускулатура, скудная растительность на лице и в подмышечных впадинах, ожирение и оволосение по женскому типу, сутулость, выраженные евнухоидные пропорции и гинекомастия (набухание грудных желез). Постоянными признаками синдрома Клайнфельтера являются недоразвитие половых органов и бесплодие.
Степень интеллектуального недоразвития у больных выражена тем глубже, чем больше дополнительных половых хромосом обнаруживается в кариотипе (46 или 49). Так, умеренная умственная отсталость зачастую приближается к психическому инфантилизму, что клинически проявляется недостаточностью внимания, восприятия, памяти, абстрактного мышления, чрезмерной внушаемостью, подражательностью, подчиняемостью, несамостоятельностью, чрезмерной привязанностью к близким, нередко с элементом назойливости. Глубокая незрелость эмоционально-волевой сферы проявляется в виде повышенного настроения, с эйфорическим оттенком, склонностью к эксплозивным аффективным вспышкам, неспособностью к длительному волевому усилию и напряженной деятельности. У больных, как правило, отсутствуют чувство долга и ответственности. При легких формах заболевания больные осознают свою неполноценность, что приводит к внутреннему конфликту и возникновению у них невротических реакций. Данным синдромом страдают лица мужского пола.
Синдром ломкой Х-хромосомы (Fragile X syndrome, FraХ). Начиная с 1980 года большое значение придают синдрому ломкой Х-хромосомы (Хq27.3) – именно с ним связывают развитие более чем 50 наследственных расстройств, включая ранний детский аутизм и 30% случаев умственной отсталости у мальчиков. Хрупкий участок Х-хромосомы впервые обнаружил Labs (1969).
Полная мутация в Х-хромосоме возникает только у женщин, и происходит это в процессе гаметогенеза, поэтому почти всегда страдают мальчики, получившие единственную Х-хромосому от матери. У девочек, получивших вторую Х-хромосому от отца, также могут быть нарушения развития, но они менее выражены, а тяжелые патологии встречаются много реже, чем у мальчиков. В отдельных случаях девочки могут получить обе ломкие хромосомы от матери, в этом случае частота и тяжесть патологии будет одинаковой с мальчиками.
Клиническую триаду синдрома ломкой Х-хромосомы образуют:
1) умеренная до степени тяжелой умственная отсталость. Лишь 30% лиц мужского пола имеют интеллект, стремящийся к нижней границе нормы, а среди женщин – носительниц такой хромосомной патологии примерно у 30% обнаруживаются признаки умственного недоразвития;
2) характерные особенности строения лица и черепа: выдающийся вперед высокий лоб, прогнатизм и удлиненные уши;
3) мальчики имеют увеличенные в размерах тестикулы (макроорхидизм).
Наблюдаются, кроме того, эпилептические припадки, синдром гиперактивности с дефицитом внимания, у более чем половины мальчиков аутизм и подобные аутизму расстройства, различные нарушения развития речи, персеверации, эхолалия, другие отклонения.
Женщины, унаследовавшие ломкую Х-хромосому с полной мутацией от своих матерей, могут быть склонны к развитию атипической депрессии, а также шизофреноподобного заболевания.
Синдром «кошачьего крика» — заболевание, обусловленное структурной аномалией 5-й пары хромосом (выпадение участка — делеция). Встречается преимущественно у девочек и характеризуется развитием умеренной или тяжелой умственной отсталости, задержкой физического развития и рядом диспластических признаков («антимонголоидный» разрез глаз, гипертелоризм, низкое расположение ушных раковин, поперечная складка ладоней и др.) Основным симптомом является своеобразный мяукающий тембр плача ребенка, связанный с аномалией строения гортани.
Синдром Вольфа—Хиршхорна.
В основе синдрома лежит изменение длины хромосомы из четвертой пары. Основные признаки заболевания у новорожденных: большое туловище, клювовидный нос и выступающее надпереносье, деформированные ушные раковины со складками, пучеглазие и колобома радужной оболочки (ее частичное отсутствие), общее недоразвитие во время беременности. Отмечается наличие четырех сгибательных складок на пальцах верхних конечностей.
Синдром Патау — комплекс врожденных пороков развития черепа, лица, нервной системы, органов слуха, зрения, внутренних органов. В основе заболевания лежит наличие добавочной хромосомы в 13-й паре. Синдром описан в 1960 г. американским педиатром Патау (К. Patau).
Клиническая картина характеризуется микроцефалией, расщелиной лица, двусторонним расщеплением верхней губы, полным расщеплением неба, маленькими глазными яблоками либо полным их отсутствием, короткой шеей, маленькими деформированными низко расположенными ушами, полидактилией, дистрофическими изменениями ногтей и костного скелета. Отмечаются также пороки развития сердца, желудка, кишечника и других органов.
Синдром трисомии-Х впервые описан в 1959 г. Частота данной патологии составляет среди новорожденных 0,1%, а среди умственно отсталых — 0,6%. Большинство лиц женского пола с трисомией-Х выявляется среди больных психиатрических лечебниц. Клиническая картина характеризуется аномалиями развития скелета, внутренних органов, различными психическими проявлениями и интеллектуальной недостаточностью. Среди полиморфизма признаков трисомии-Х наиболее характерными являются: низкий рост, аномалии ушей, прикуса, высокое стояние твердого неба, короткие пальцы, искривленный мизинец, широкий промежуток между 1 и 2 пальцами на стопах, синдактилия, недоразвитие половых функций.
Умственная отсталость проявляется в виде легкой или умеренной степени. Характерны эмоциональные расстройства (вспыльчивость, агрессивность, неустойчивость настроения и немотивированные поступки). Девочки с синдромом трисомии-Х с трудом, но в большинстве случаев (легкая степень умственной отсталости) обучаются в массовых школах.
Синдром Эдвардса — наследственное заболевание, обусловленное, как правило, трисомией 18-й хромосомы и проявляющееся множественными пороками развития органов и систем. Синдром описан в 1960 г. американским педиатром Эдвардсом (J. Edwards).
Клиническая картина заболевания характеризуется задержкой психического развития, множественными аномалиями лица, костно-мышечной системы, черепа и головного мозга.
К хромосомным синдромам, помимо вышеописанных, относится большая группа так называемых семейных форм умственной отсталости, когда совершенно точно доказано наличие данной патологии у близких родственников.
Синдром Аперта (акроцефалосиндактилия) — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, экзофтальмом, деформацией зубов и синдактилиями. Синдром описан французским педиатром Апертом (Е. Apert) в 1906 г.
Синдром Крузона — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, преждевременным срастанием швов черепа, уменьшением мозгового вещества, экзофтальмом, вторичной атрофией зрительных нервов, прямоугольным расположением большого пальца к кисти. Впервые синдром описан французским врачом Крузоном (О. Crouson) в 1912 г.
Синдром Сьегрена—Ларссона — наследственное заболевание, которое сопровождается умственной отсталостью, парезами (снижением силы) конечностей и ухудшением зрения.
Синдром Берьесона—Форсмана—Лемана — синдром характеризующийся умственной отсталостью в сочетании с избыточным весом. Впервые описан американскими врачами Берьесоном (М. Berjeson) Форсманом (Н. Foreman) и Леманом (О. Lehman) в 1963 г. Клиническая картина заболевания проявляется выраженным ожирением и прогрессирующей умственной отсталостью. Ожирение носит не равномерный характер. Жир откладывается преимущественно на бедрах, груди и лице, что придает своеобразный вид такому больному (бочкообразная карликовая фигура с заплывшим лицом, большими ушами и узкими разрезами глаз). У больных часто отмечаются эпилептические припадки. Умственная отсталость колеблется от умеренной до тяжелой степени. Данная патология встречается только у лиц мужского пола, но носителями патологического гена являются женщины.
Синдром Прадера—Вилли — наследственное заболевание, характеризующееся глубокой умственной отсталостью, низким ростом, гипогенитализмом, ожирением, резко выраженной мышечной гипотонией.
Синдром Книппеля—Фейля (синдром короткой шеи) — наследственное семейное заболевание, обусловленное врожденными аномалиями развития скелета и внутренних органов в сочетании с тяжелой степенью умственной отсталости. Клиника синдрома подробно описана французскими врачами Клиппелем Фейлем в 1912 г.
Аномалия развития характеризуется следующими проявлениями: короткой шеей как результат количественного уменьшения шейных позвонков, ограничением подвижности головы, расщеплением твердого неба, бочкообразной грудной клеткой, врожденными пороками сердца, добавочными долями или отсутствием отдельных долей легких, синдактилиями (сращение пальцев конечностей), глухотой вследствие заращения наружных слуховых проходов, сужением анального отверстия и многими другими симптомами. Интеллектуальная недостаточность является результатом тяжелой умственной отсталости
Лечение ЗПРР при хромосомных заболеваниях.
Основой лечения является уникальная методика патогенетической терапии речевых расстройств при хромосомной патологии — биофизическая активация нейромоторных структур, основу которого составляет щадящая стимуляция проводников нервной системы микротоками с использованием нейрофизиологического прибора. Метод лечения базируется как на активации самих речевых центров, так и на восстановлении нарушенных связей между центрами и полушариями головного мозга. Помимо этого, восстанавливаются разрозненные связи речевых центров с другими областями мозга, участвующими в реализации речевой функции. В процессе лечения формируется физиологичное, последовательное взаимодействие всех зон мозга, связанных с речепродукцией. В результате появляется речь.
Проведение биофизической активации сочетается с дополнительными методиками лечения, такими как — лимфомежклеточная терапия, которая применяется для регулирования интегративной деятельности и восполнения дефицита энергетической системы мозга и позволяющая применять малые дозы церебропротекторов, которые вводятся эндолимфатически и попадают в ткани головного мозга, минуя гематоэнцефалический барьер.
В качестве другого способа использования препаратов с нейротрофическим и антиоксидантным действием применяется методика эндоназального электрофореза кортексина, что позволяет вводить лекарственные препараты непосредственно в ткани головного мозга.
Исследования последних десятилетий выявили, что у большинства детей с речевыми и поведенческими проблемами в различной степени нарушены функции мозжечка и базальных ганглиев. Именно функционирование мозжечка определяет успешность ребенка в обучении. С этой целью применяется уникальная разработка Центра авиакосмической медицины — подошвенный имитатор опорной нагрузки «Корвит», применяемый для нейрофизиологической регуляции стато-кинетической функции ЦНС. В основе терапевтического воздействия аппарата «Корвит» лежит процесс активации опорной афферентации, отвечающей за нормализацию процессов возбуждения и торможения в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в головном мозге, способствующих восстановлению координации движений, и, опосредованно, улучшению речи и мышления.
Также для успешного лечения различных форм ЗПРР специалистами применяется одно из достижений современной науки — метод аудиовокальной терапии RUSTOMATIS. Прибор использует звукозаписи высокочастотных и низкочастотных компонентов. При чередовании такой музыки путем напряжения и расслабления у ребенка тренируется аппарат среднего уха – молоточек и стремечко, с помощью чего расширяется диапазон восприятия внешних факторов, увеличивается концентрация внимания, в мозг поступает новая информация и, как следствие исчезают многие нарушения и расстройства.
Обязательным звеном в лечебном комплексе у детей с наличием речевых расстройств является занятия с клиническим психологом, а также логопедическая коррекция, которая включает диагностику степени нарушений, ежедневные занятия, направленные на улучшение речевой функции и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
На фоне сочетания проведения биофизической активации со вспомогательными методиками лечения наблюдаются положительные изменения, которые могут быть видны уже через несколько процедур, но максимальный эффект развивается через полтора-три месяца после курса. Как правило, для закрепления полученных результатов и дальнейшего развития двигательных и когнитивных навыков специалистами центра рекомендуется повторный курс лечения через 5-6 месяцев.
Синдром кошачьего крика
⇐ ПредыдущаяСтр 2 из 2
болезнь кошачьего крика, синдром Лежена — по имени описавшего его в 1963 году французского учёного — редкое генетическое расстройство, вызываемое отсутствием фрагмента 5-й хромосомы.
Кариотип 46 XX (ХУ), 5р-.
Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом.
Хромосомно синдром кошачьего крика объясняется частичной моносомией; он развивается при делеции (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы.
При этом синдроме наблюдается:
· общее отставание в развитии,
· низкая масса при рождении и мышечная гипотония,
· лунообразное лицо с широко расставленными глазами,
· характерный плач ребёнка, напоминающий кошачье мяуканье, причиной которого является изменение гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани. Признак исчезает к концу первого года жизни.
Частота синдрома примерно 1:45000.
Клиническая картина синдрома и продолжительность жизни людей с этим синдромом довольно сильно варьируются по сочетанию врождённых пороков развития органов.
Аутосомные трисомии.
Синдром Дауна.
Синдром Дауна был описан в 1866 году английским педиатром Л. Дауном, но только в 1959 г. французским генетиком и врачом Дж. Леженом было доказано, что это заболевание хромосомной природы, а именно – трисомия по хромосоме 21.
Частота – 1:700-800 новорожденных, одинаково часто наблюдается у обоих полов.
Кариотип – 47,ХХ(ХY)+21.
Для больных характерны следующие признаки:
Округлой формы голова с уплощенным затылком,
Лоб скошенный и узкий,
Лицо плоское, типичен эпикант, плоская спинка носа,
Монголоидный разрез глаз,
Постоянно открытый рот, толстые губы,
Большой складчатый язык,
Двух-фаланговый мизинец.
Для всех больных этим синдромом характерна умственная отсталость. Среди детей без врожденных пороков развития дебильность отмечается у 75% детей, имбицильность – у 20%, идиотия у 5%.
Средняя продолжительность жизни больных ниже, чем в популяции. В последние десятилетия имеется постоянная тенденция к увеличению продолжительности жизни больных с синдромом Дауна. В настоящее время есть описания больных старше 70 лет.
Прогноз жизни определяется, в основном, наличием пороков развития сердечно-сосудистой и пищеварительной систем. Хотя на первом году жизни дети часто погибают от пневмоний и острого лейкоза, что связано с несостоятельностью их иммунной системы.
Хорошо известно, что дети с этим синдромом чаще рождаются у матерей старше 35 лет. Причины такой зависимости до конца не ясны. Около 80% всех случаев обусловлено аномальными яйцеклетками и лишь 20% аномальными сперматозоидами.
Синдром Патау.
Генетическая природа этого синдрома трисомия 13была расшифрована в 1960 г. американским генетиком К.Патау.
Частота 1:6000, занимает второе место после синдрома Дауна (среди аутосомных трисомий).
Кариотип – 47,ХХ(ХY)+13.
Мальчики и девочки страдают с одинаковой частотой.
Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией. Средняя масса при рождении 2500гр., что на 900гр. меньше нормы.
Фенотипические признаки синдрома настолько характерны, что позволяют практически сразу заподозрить это заболевание:
Аномалии черепа и лица- микроцефалия, скошенный лоб,
Узкие глазные щели, запавшее переносье,
Низко расположенные и деформированные ушные раковин.
Наиболее характерными пороками развития являются:
расщелины губи и неба и полидактилия.
Продолжительность жизни резко снижена. На первом году жизни умирает 95% больных, в возрасте старше 3-х лет остаются в живых единицы. Все дети с синдромом Патау имеют тяжелую умственную отсталость (глубокая идиотия).
Синдром Эдвардса.
Этот синдром был описан в 1960г. Д.Эдвардсом, его генетическая природа – трисомия 18.
Частота 1:7000. Девочки поражаются в 3 раза чаще, причины этого неизвестны.
Фенотипические проявления довольно характерны:
Череп долихоцефалической формы,
Нижняя челюсть и отверстие рта маленькие,
⇐ Предыдущая12
Поиск по сайту:
Синдром кошачьего крика
Синдром кошачьего крика также известен как делеции 5р-синдром, частичной делеции короткого плеча 5-й хромосомы синдром, 5р-синдром.
Основным дефектом является частичная (как терминальная так и интерстициальная) делеция 5р15.2 — р15.3. Делеция может быть результатом мутации «de novo» (в 85%) или же наследоваться от родителей — носителей несбалансированной транслокации (в 15%). В этом случае проявления синдрома кошачьего крика тяжелее.
Делеции, захватывающие проксимальный участок, не вызывают характерного для синдрома кошачьего крика фенотипа. Отмечено прямую зависимость между размером делеции и проявлениями синдрома кошачьего крика, в частности, чем больше делеция, тем тяжелее степень микроцефалии, умственной отсталости и пренатальной гипотрофии.
Синдром кошачьего крика диагностируется у около 1% лиц с умственной отсталостью, которые находятся в специализированных учреждениях. При специальных занятиях и в благоприятных условиях окружения, некоторые дети достигают психомоторного развития здорового 5-6-летнего ребенка. Половина детей старше 10 лет имели словарный запас, достаточный для общения (Wilkins at al, исследования группы 65 детей с синдромом кошачьего крика).
Высокого тембра пронзительный крик характерен для детей с этим заболеванием в грудном возрасте, хотя не всегда и не у всех пациентов он наблюдается. Некоторые авторы предполагают, что этот своеобразный крик центрального происхождения и не связан с аномалией гортани. С возрастом ребенка он постепенно исчезает, что иногда затрудняет диагностику данной патологии у пациентов старшего возраста.
Со временем лицо становится асимметричным, округлость исчезает.
Характерны частые инфекции верхних дыхательных путей, отиты, проблемы с вскармливанием.
Аномалии, соединенные с синдромом кошачьего крика:
Общие:
- Пренатальная гипотрофия (<2500 г) — 72%.
- Задержка физического развития — 100%.
- Плач, подобный кошачьего крика, примерно — 100%.
- Умственная отсталость — 100%.
- Гипотония — 78%.
Черепно-лицевые:
- Микроцефалия — 100%.
- Круглое лицо — 68%.
- Эпикант — 85%.
- Опущенные углы глазных щелей — 81%.
- Косоглазие, чаще расходятся — 61%.
- Низкорасположенным диспластические уши — 58%.
Сердце:
- Врожденные пороки сердца (различные типы) — 30%.
Верхние конечности:
- 4-х пальцевая ладонная складка — 81%.
- Дистальный аксиальный трирадиус — 40%.
Другие аномалии:
- Расщелины губы и неба, миопия, атрофия зрительного нерва, преаурикулярные папилломы, расщелины язычка, аномалии прикуса, короткая шея, клинодактилия, паховая грыжа, крипторхизм, отсутствие почек и селезенки, полупозвонки, плоскостопие, сколиоз, раннее поседение волос.
Частота
Частота возникновения синдрома кошачьего крика составляет около 1 на 50000 рождений.
Диагностика
Пренатальная диагностика синдрома кошачьего крика базируется на использовании FISH-метода в случае, если женщина является носителем сбалансированной транслокации.
Антенатальные ультразвуковые признаки синдрома кошачьего крика
Поскольку синдром кошачьего крика встречается в среднем с частотой 1: 50000, описано всего несколько случаев антенатальных ультрасонографических находок при синдроме кошачьего крика.
Ультразвуковое обследование 26-летней первобеременной, срок гестации 19 недель, направленное по поводу подозрения на водянку плода. При УЗ-обследовании выявлено анасарка, выпот в плевральную полость плода, асцит, гипоплазию мозжечка, дефект межжелудочковой перегородки больших размеров, аорту, укорочение указательных пальцев. Был обнаружен кистозный творение шеи размером 12 на 18 мм, без перегородок. Бипариетальный диаметр и длина бедра соответствовали срока гестации 19 недель. При проведении пульсовой допплерографии выявлено отсутствие диастолического кровотока в средней мозговой артерии и обратный кинцеводиастоличный кровоток в артерии пуповины, что расценивалось как признак наступающей гибели плода. Через два дня была диагностирована внутриутробная гибель плода и проведены индуцированные вагинальные роды. Родился плод женского пола, весом 600 граммов. При патанатомичном исследовании пренатальный ультразвуковой диагноз был полностью подтвержден: выявлено анасарка, асцит, выпот в плевральной полости, гипоплазию мозжечка, большой дефект межжелудочковой перегородки. Кистозное творение было локализировано подкожно, в области затылочной шейной складки, с прозрачным серозным содержимым, перепонок не было. Гистологически — стенка кисты образована слоем цилиндрических клеток. При цитогенетическом обследовании выявлено частичную делецию короткого плеча 5-й хромосомы. Гистологическое исследование плаценты патологических изменений не выявило.
Таким образом, в данном случае при пренатальном ультразвуковом обследовании было выявлено гипоплазию мозжечка, большой дефект межжелудочковой перегородки, аномалия аорты и кистозный образование в области шейной складки, впоследствии подтвердилось при аутопсии.
Было описано несколько случаев синдрома кошачьего крика, при которых наблюдалась эдема затылочной шейной складки и ее кисты. Примерно 35% плодов, у которых диагностирована кистозная гигрома шеи имеют кариотип 45 ХО и другие хромосомные аномалии, среди которых — трисомия 21 и 18. При антенатальном выявлении кистозной гигромы шеи и гипоплазии мозжечка можно заподозрить анеуплоидии у плода, а также, исходя из описанных случаев — другие хромосомные аномалии, в частности частичную делецию 5-й хромосомы.
Синдром делеции короткого плеча хромосомы
Синдром делеции короткого плеча хромосомы 4 синонимы… [Стр.203]
СИНДРОМ ДЕЛЕЦИИ КОРОТКОГО ПЛЕЧА ХРОМОСОМЫ 5 синонимы… [Стр.523]
Частичная делеция короткого плеча хромосомы 5 (5р-) (синдром кошачьего крика см. т. 1, гл. 30)… [Стр.577]
| Рис. 217. Синдром кошачьего крика (Сп-би-сба. (А) — пациент с синдромом кошачьего крика, (Б) — кариотип ребенка с синдромом кошачьего крика 46 ХУ(5р-). Стрелкой указана делеция в коротком плече хромосомы 5… |
Синдром, связанный с делецией короткого плеча хромосомы 4, выделен в 1965 г. независимо исследователями — немецкими генетиками во главе с U. W и американскими, возглавляемы-… [Стр.408]
Диагноз синдром аниридия — опухоль Внльмса . При цитогенетическом исследовании обнаружена интерстициальная делеции короткого плеча хромосомы 11-46,XX, (И) (Р11,2-р 15.1). [Стр.419]
Дифференциальная диагностика. Трисомия 22 и делеция короткого плеча хромосомы 9 могут клинически напоминать синдром Дауна, что требует обязательного исследования кариотипа. [Стр.412]
Делеция короткого плеча хромосомы 4 (синдром 4р, или синдром Вольфа—Хиршхорна). Впервые эта делеция была описана в 1965 г. Ее клинические проявления приведены в табл. 8.10. [Стр.195]
При цитогенетической верификации синдрома выявляется либо простая делеция короткого плеча 4-й хромосомы (4 р—), либо делеция, замаскированная транслоцированным фрагментом (см. синдром Эдвардса), либо кольцевая хромосома 4. [Стр.162]
Синдром кошачьего крика (C .) — обусловлен частичной делецией короткого плеча 5-й хромосомы и сочетается с низко расположенными ушами и пороком сердца. [Стр.118]
Синдром моносомии 9р (синдром Альфи) впервые описан Альфи и соавт. в 1973 г. Он возникает вследствие частичных делеций короткого плеча хромосомы 9, изохромосом по длинному плечу и несбалансированных трансло-… [Стр.585]
Синдром кошачьего крика — наследственное заболевание, определяемое делецией короткого плеча пятой хромосомы человека. [Стр.277]
Смотреть другие источники с термином Синдром делеции короткого плеча хромосомы:
[Стр.485]
[Стр.841]
[Стр.204]
[Стр.410]
[Стр.415]
[Стр.417]
[Стр.16]
[Стр.652]
[Стр.110]
[Стр.485]
[Стр.841]
[Стр.204]
[Стр.410]
[Стр.415]
[Стр.417]
[Стр.16]
[Стр.652]
[Стр.110]
[Стр.685]
[Стр.162]
[Стр.28]
[Стр.358]
[Стр.358]
[Стр.361]
[Стр.38]
[Стр.519]
[Стр.583]
[Стр.33]
[Стр.230]
[Стр.214]
[Стр.172]
[Стр.150]
[Стр.27]
[Стр.23]
[Стр.97]
[Стр.181]
[Стр.196]
[Стр.671]
[Стр.682]
[Стр.164]
[Стр.485]
[Стр.111]
[Стр.39]
[Стр.39]
[Стр.65]
| От 80% до 99% людей имеют эти симптомы | ||
| Кошачий крик | кошачий крик | 0200046 |
| Эпикантус | Складки глаз Выраженные складки глаз [ более ] | 0000286 |
| Высокий голос | 0001620 | |
| Умственная отсталость тяжелая | Ранняя и тяжелая умственная отсталость Умственная отсталость, тяжелая форма Тяжелая умственная отсталость [ более ] | 0010864 |
| Низко посаженные, повернутые назад уши | 0000368 | |
| Микроцефалия | Аномально маленький череп Уменьшение окружности черепа Уменьшенный размер черепа Уменьшенная окружность головы Небольшая окружность головы [ более ] | 0000252 |
| Microretrognathia | Маленький втянутый подбородок | 0000308 |
| Мышечная гипотония | Низкий или слабый мышечный тонус | 0001252 |
| Круглый торцевой | Круглое лицо Круглый внешний вид лица Круглая форма лица [ более ] | 0000311 |
| Серьезная задержка глобального развития | 0011344 | |
| Широкая переносица | Широкая носовая перемычка Широкий корень носа Расширенный носовой мост Увеличенная ширина переносицы Увеличенная ширина переносицы Увеличенная ширина переносицы Увеличенная ширина переносицы Носовая перемычка широкая Широкая переносица Расширенный носовой мост [ более ] | 0000431 |
| 30% -79% людей имеют эти симптомы | ||
| Нисходящие глазные щели | Наклон отверстия между веками вниз | 0000494 |
| Высокое небо | Повышенное небо Увеличенная небная высота [ более ] | 0000218 |
| Гипертелоризм | Широко посаженные глаза Широко расставленные глаза [ более ] | 0000316 |
| Задержка внутриутробного развития | Дефицит дородового роста Задержка внутриутробного развития [ более ] | 0001511 |
| Сколиоз | 0002650 | |
| Короткая шея | Уменьшенная длина шеи | 0000470 |
| Низкий рост | Уменьшенный рост Маленький рост [ более ] | 0004322 |
| Маленькая рука | Непропорционально маленькие руки | 0200055 |
| 5% -29% людей имеют эти симптомы | ||
| Нарушение минеральной плотности кости | 0004348 | |
| Нарушение морфологии сердечно-сосудистой системы | 0030680 | |
| Синдактилия пальца | 0006101 | |
| Паховая грыжа | 0000023 | |
| Гипергибкость суставов | Суставы выходят за пределы ожидаемого диапазона движений | 0005692 |
| Преаурикулярная кожная бирка | 0000384 | |
| Рецидивирующие переломы | Повышенная частота переломов Повышенные переломы Множественные переломы Множественные самопроизвольные переломы Различная степень множественных переломов [ более ] | 0002757 |
| Процент людей, у которых есть эти симптомы, недоступен через HPO | ||
| Патология почек | Аномальная почка | 0000077 |
| Аномалия ушной раковины | Уши неправильной формы Порок развития ушной раковины Деформированные уши Деформированные уши [ более ] | 0000377 |
| Агрессивное поведение | Агрессия Агрессивное поведение Агрессивность [ более ] | 0000718 |
| Нарушение прикуса переднего открытого прикуса | Отсутствие перекрытия передних верхних и нижних зубов Промежуток между верхними и нижними передними зубами при прикусывании [ более ] | 0009102 |
| Беспокойство | Чрезмерное, постоянное беспокойство и страх | 0000739 |
| Аутизм | 0000717 | |
| Двустворчатый язычок | 0000193 | |
| Катаракта | Помутнение хрусталика глаза Мутный объектив [ более ] | 0000518 |
| Довольно счастливый характер | 0100024 | |
| Крипторхизм | Неопустившиеся яички Неопустившееся яичко [ более ] | 0000028 |
| Задержка речевого и языкового развития | Нарушение речевого развития Задержка языкового развития Отложенная речь Задержка получения речи Задержка речевого развития Нарушение речи и языкового развития Нарушение речевого развития Языковая задержка Язык отложен Дефицит языкового развития Позднее развитие речи Плохое языковое развитие Задержка речи и языка Речевые и языковые трудности Задержка речи [ более ] | 0000750 |
| Диастаз прямых мышц живота | Разрыв между большими левой и правой мышцами живота | 0001540 |
| Затруднения при ходьбе | Трудности при ходьбе | 0002355 |
| Углы рта опущенные | Опущенные уголки рта Опущенный рот [ более ] | 0002714 |
| Эхолалия | Повторение речи другого человека | 0010529 |
| Асимметрия лица | Асимметрия лица Кривое лицо Несимметричное лицо [ более ] | 0000324 |
| Гримаса | 0000273 | |
| Проблемы с кормлением в младенчестве | 0008872 | |
| Функциональные респираторные нарушения | 0002795 | |
| Гастроэзофагеальный рефлюкс | Кислотный рефлюкс Кислотная рефлюксная болезнь Изжога [ более ] | 0002020 |
| Задержка роста | Задержка роста Дефицит роста Нарушение роста Задержка роста Плохой рост Замедленный рост [ более ] | 0001510 |
| Нарушение слуха | Глухота Дефект слуха [ более ] | 0000365 |
| Большой осевой трирадиус | 0001042 | |
| Гиперактивность | Более активен, чем обычно | 0000752 |
| Гиперакузис | 0010780 | |
| Гипертония | 0001276 | |
| Гипоспадия | 0000047 | |
| Умственная отсталость | Умственная отсталость Умственная отсталость Умственная отсталость неспецифическая Умственная отсталость [ более ] | 0001249 |
| Длинная поверхность | Удлинение лица Увеличенная высота лица Увеличенная длина лица Вертикальное удлинение лица Вертикальное увеличение лица Вертикальное разрастание лица [ более ] | 0000276 |
| Низко посаженные уши | Низко посаженные уши Низко посаженные уши [ более ] | 0000369 |
| Приводная мышца плюсны | Передняя половина стопы поворачивается внутрь | 0001840 |
| Близорукость | Близкий вид Близорукий Близорукость Близорукость [ более ] | 0000545 |
| Узкое лицо | Уменьшение ширины лица Уменьшение ширины лица [ более ] | 0000275 |
| Гипотония новорожденных | Низкий мышечный тонус в неонатальном периоде | 0001319 |
| Вызывающее оппозиционное расстройство | 0010865 | |
| Атрофия зрительного нерва | 0000648 | |
| Чрезмерное дружелюбие | 0100025 | |
| Pes planus | Плоскостопие Плоскостопие [ более ] | 0001763 |
| Преждевременное поседение волос | Раннее поседение Преждевременное поседение Преждевременное поседение Преждевременное поседение волос [ более ] | 0002216 |
| Выступающие надглазничные гребни | Выдающаяся бровь | 0000336 |
| Рецидивирующие инфекции в младенчестве и раннем детстве | 0005437 | |
| членовредительство | Умышленное членовредительство Членовредительство [ более ] | 0000742 |
| Короткая продолжительность концентрации внимания | Плохая концентрация внимания Проблема с вниманием [ более ] | 0000736 |
| Короткая пястная кость | Укороченная длинная кость руки | 0010049 |
| Короткая плюсневая кость | Короткая длинная кость стопы | 0010743 |
| Короткий желобок | 0000322 | |
| Одиночная поперечная ладонная складка | 0000954 | |
| Малый для гестационного возраста | Вес при рождении менее 10-го процентиля Низкий вес при рождении [ более ] | 0001518 |
| Спорадически | Нет предыдущего семейного анамнеза | 0003745 |
| Стеноз наружного слухового прохода | Сужение прохода от внешнего к среднему уху | 0000402 |
| Стереотипия | Повторяющиеся движения Повторяющееся или самоповреждающее поведение [ более ] | 0000733 |
| Косоглазие | Косоглазый Косоглазие Прищуренные глаза [ более ] | 0000486 |
| Синдактилия | Перепончатые пальцы рук или ног | 0001159 |
| Киноварь толстой нижней губы | Увеличение объема нижней губы Пухлая нижняя губа Выступающая нижняя губа [ более ] | 0000179 |
Обзор синдрома Кри дю Чат
Синдром Кри дю Чата (по-французски «кошачий крик») — редкое хромосомное заболевание, вызванное отсутствием или удалением частей хромосомы 5.Младенцы, рожденные с этим синдромом, часто издают пронзительный крик, похожий на кошачий, отсюда и название состояния. Поскольку это состояние возникает из-за отсутствия частей короткого плеча (p) хромосомы 5, Cri du Chat также известен как синдром 5p- (5p минус).
Дмитрий Отис / Getty Images
Симптомы
Ключевые физические характеристики и симптомы синдрома Кри-дю-Шат вызваны отсутствием или удалением генов в маленьком плече (p) хромосомы 5. Исследователи подозревают, что конкретный набор симптомов, связанных с Кри-дю-Шат, и тяжесть этих симптомов, связано с размером и расположением удаленной или отсутствующей части хромосомы.
Как и при других хромосомных нарушениях, симптомы и тяжесть состояния варьируются от человека к человеку. Однако есть несколько ключевых проявлений этого состояния, которые заметны с рождения. Эти отличительные особенности включают в себя:
- Низкая масса тела при рождении
- Слабый сосательный рефлекс
- Медленный рост или отсутствие роста
- Пронзительный, мяукающий крик, похожий на кошачий
- Низкий мышечный тонус
Хотя они могут не обладать всеми функциями, многие новорожденные с Cri du Chat имеют отличительные физические характеристики, в том числе:
- Маленькая голова (микроцефалия) и челюсть
- Аномально круглое лицо
- Неправильный прикус зубов
- Широко посаженные, наклоненные вниз глаза
- Лишние кожные складки вокруг глаз
- Низко посаженные уши
- «Тесьма» пальцев рук и ног (синдактилия)
- Расщелина губы или неба
По мере того как дети с этим заболеванием вырастают, они могут начать проявлять и испытывать спектр симптомов, связанных с Cri du Chat, а также другие расстройства, обычно обнаруживаемые у людей с этим заболеванием, в том числе:
- Задержка моторики, когнитивных функций и речи
- Умственная отсталость от умеренной до тяжелой
- Психомоторная инвалидность
- Изъятия
- Аутизмоподобное поведение, такое как взмахи руками, покачивание и чувствительность к шуму
- Сколиоз
- Врожденные пороки сердца (около 15–20 процентов пациентов)
- Грыжи
- Поведенческие проблемы, такие как истерики и плохое внимание / контроль над импульсами
- Медленная, осторожная походка или необходимость использования средств передвижения, включая инвалидные коляски
- Саморазрушительное поведение, такое как битье головой и выдергивание кожи
- Рецидивирующие инфекции (особенно респираторные, ушные и желудочно-кишечные)
- Близорукость
- Запор
- Нарушения со стороны почек или мочевыводящих путей
- Преждевременное поседение волос
- Проблемы со сном
- Проблемы приучения к туалету
Причины
Синдром Кри дю Шат был впервые описан в 1963 году французским педиатром Жеромом Леженом.Лежен наиболее известен тем, что открыл генетическую основу трисомии 21 (синдрома Дауна).
Заболевание считается очень редким — в США ежегодно рождается около 50-60 младенцев с синдромом Cri du Chat. Заболевание чаще поражает женщин, чем мужчин, и диагностируется у людей любого этнического происхождения.
Хотя Cri du Chat связано с генами, это не обязательно наследственное заболевание. Большинство случаев происходит de novo (или спонтанно) во время эмбрионального развития.Исследователи не уверены, почему происходят эти удаления. Родители ребенка, рожденного с кри дю Шат из-за спонтанной делеции, будут иметь нормальные хромосомы. Поэтому, если в будущем у них будет еще один ребенок, маловероятно, что еще один ребенок родится с этим заболеванием.
В некоторых случаях заболевание возникает из-за того, что гены перемещаются с одной хромосомы на другую. Это приводит к перестройке генетического материала. Транслокации между хромосомами могут происходить спонтанно или передаваться по наследству от родителя, который является носителем пораженного гена.
Исследователи подозревают, что у людей с Cri du Chat, у которых есть серьезная умственная отсталость, могут быть делеции в определенном гене CTNND2. Необходимы дополнительные исследования потенциальной связи между симптомами состояния и конкретными генами, но по мере того, как больше узнается о взаимосвязи, вероятно, будет лучше понятна причина. Понимание того, почему происходят делеции в гене, будет важной частью постановки диагноза и лечения для улучшения жизни людей с Cri du Chat.
Диагностика
Большинство случаев Cri du Chat можно диагностировать при рождении в рамках тщательного обследования новорожденного. Ключевые физические особенности этого состояния, особенно микроцефалия, легко идентифицируются у новорожденных. Другие сопутствующие симптомы, такие как «кошачий плач» ребенка, низкий мышечный тонус и плохой сосательный рефлекс, также проявляются вскоре после рождения.
Несколько различных типов генетического тестирования, включая кариотипирование, флуоресцентную гибридизацию in situ (FISH) и анализ хромосомных микрочипов, можно использовать для поиска делеций в хромосоме 5, которые являются диагностическими для Cri du Chat.
Врач также может назначить более узкоспециализированные тесты, чтобы определить, были ли делеции спонтанными или из-за пораженного гена родителя. В последнем случае существуют тесты, которые могут определить, у какого из родителей есть транслоцированный ген.
Растущая доступность более специализированных методов генетического тестирования позволила диагностировать некоторые случаи Cri du Chat пренатально.
Лечение
Серьезность Cri du Chat бывает разной.Опыт наличия, постановки диагноза и лечения этого состояния и его симптомов будет уникальным для каждого человека, у которого оно есть. Семьи, у которых есть дети с Cri du Chat, часто обращаются за помощью к разным поставщикам медицинских услуг, включая смежных медицинских работников, социальных работников и специалистов в области образования. После рождения ребенка с Cri du Chat родители обычно направляются на генетическую консультацию.
Поскольку Cri du Chat часто диагностируется при рождении или вскоре после этого, семьи могут сразу же приступить к созданию группы поддержки.Раннее вмешательство помогает семьям разработать стратегии управления физическими и эмоциональными различиями, с которыми сталкиваются дети с кри дю Шат по сравнению со своими сверстниками.
Большинство детей с диагнозом Cri du Chat начинают лечение до своего первого дня рождения. Это часто включает комбинацию физической, профессиональной и логопедической терапии. Если у пациента есть сопутствующее заболевание, такое как врожденный порок сердца, ему также потребуются более специализированные медицинские услуги в дополнение к обычному уходу.
Родителям может потребоваться поискать общественные и академические ресурсы, чтобы помочь детям с Cri du Chat приспособиться к школе. Программы специального образования являются одним из вариантов, зависящих от типа и степени обучаемости и / или физических недостатков ребенка, а также с учетом их социальных и поведенческих потребностей. Некоторые семьи предпочитают обучать детей на дому с помощью Cri du Chat или записывать их в специально разработанные школы или программы.
На ожидаемую продолжительность жизни пациентов с Cri du Chat это состояние не влияет напрямую, хотя осложнения, связанные с его особенностями, например развитие аспирационной пневмонии, если они склонны к респираторным проблемам, могут представлять риск.Во многих задокументированных случаях дети с Cri du Chat дожили до среднего возраста и старше. Однако люди, у которых есть Cri du Chat, не всегда могут жить самостоятельно. Многим взрослым с этим заболеванием потребуются поддерживающие медицинские, социальные, индивидуальные и профессиональные услуги.
Альтернативные и дополнительные методы лечения также могут быть полезны для пациентов с Cri du Chat, особенно в детском и подростковом возрасте. Игровая терапия, ароматерапия, музыкальная терапия и терапия с участием животных — все это приносит пользу детям с помощью Cri du Chat.
Детям с более тяжелыми формами инвалидности требуются зонды для кормления (парентеральное питание), а тем, кто ведет серьезные самоповреждения, может потребоваться дополнительный уход. Медсестры на дому, общежития или учреждения для престарелых также являются вариантами для семей, которые нуждаются в помощи, чтобы помочь своему ребенку жить полноценной, безопасной, счастливой и здоровой жизнью.
Слово Verywell
Симптомы Cri du Chat различаются по спектру и могут включать тяжелые умственные и физические нарушения, задержку речи или двигательных функций, а также поведенческие проблемы или другие заболевания, такие как врожденные пороки сердца или сколиоз.Осложнения, связанные с этим заболеванием, или те, которые обычно сочетаются с ним, могут вызвать серьезные проблемы со здоровьем, но важно помнить, что большинство людей с Cri du Chat доживают до среднего возраста и старше.
Синдром Кри дю Шат | Orphanet Journal of Rare Diseases
Lejeune J, Lafourcade J, Berger R, Vialatte J, Boeswillwald M, Seringe P, Turpin R: Trois cas de délétion partielle du bras court d’un хромосома 5. CR Acad Sci (D ). 1963, 257: 3098-3102.
CAS
Google Scholar
Overhauser J, Huang X, Gersh M, Wilson W., McMahon J, Bengtsson U, Rojas K, Meyer M, Wasmuth JJ: Молекулярное и фенотипическое картирование короткого плеча хромосомы 5: сублокализация критической области для синдрома кри-дю-чат. Hum Mol Genet. 1994, 3: 247-252.
CAS
PubMed
Google Scholar
Симмонс А.Д., Гудард С.А., Галлардо Т.Д., Оверхаузер Дж., Ловетт М.: пять новых генов из критической области кри-дю-чат, выделенных прямым отбором.Hum Mol Genet. 1995, 4: 295-302.
CAS
PubMed
Google Scholar
Хигураши М., Ода М., Иидзима К., Иидзима С., Такешита Т., Ватанабэ Н., Йонеяма К.: Распространенность живорождений и последующее наблюдение синдромов пороков развития у 27 472 новорожденных. Brain Dev. 1990, 12: 770-773.
CAS
PubMed
Google Scholar
Нибур Э: Синдром кри-дю-чата. Эпидемиология, цитогенетика и клинические особенности.Hum Genet. 1978, 44: 227-275. 10.1007 / BF00394291.
CAS
PubMed
Google Scholar
Duarte AC, Cunha E, Roth JM, Ferriera FL, Garcias GL, Martino-Roth MG: Цитогенетика генетического консультирования пациентов в Пелотасе, Риу-Гранди-ду-Сул, Бразилия. Genet Mol Res. 2004, 3: 303-308.
CAS
PubMed
Google Scholar
Dallapiccola B: Malattia del «cri du chat» (5p-).La patologia cromosomica — Atti dei Congressi della Società Italiana di Medicina Interna, 74 ° Congresso, Montecatini, 21–24 октября. 1973, Рома: Л. Поцци, 416-436.
Google Scholar
Даллапиккола Б., Писточчи Г., Форабоско А., Капра Л.: Изменения скелета при синдроме «кри-дю-чат». Acta Genet Med Gemellol. 1973, 22: 39-44.
CAS
PubMed
Google Scholar
Cerruti Mainardi P, Vianello MG, Bonioli E: Рассмотрите вопрос о 5 casi di sindrome di «cri du chat». Минерва Педиатр. 1976, 28: 2389-2400.
CAS
PubMed
Google Scholar
Wilkins LE, Brown JA, Nance WE, Wolf B: Клиническая неоднородность у 80 детей, воспитываемых дома, с синдромом кри-дю-чат. J Pediatr. 1983, 102: 528-533. 10.1016 / S0022-3476 (83) 80179-6.
CAS
PubMed
Google Scholar
Schinzel A: Каталог несбалансированных хромосомных аберраций у человека Берлин: Вальтер де Грюйтер; 1984.
Google Scholar
Benigno V, Cammarata M, Giuffrè L: La sindrome del «cri du chat»: dermatoglifi palmari di interesse diagno. Минерва Педиатр. 1985, 37: 251-253.
CAS
PubMed
Google Scholar
Fenger K, Niebuhr E: Дискриминантный анализ дерматоглифических паттернов подошвы и ладони у датских критических пробандов и нормальной контрольной группы.J Ment Defic Res. 1985, 29: 281-288.
PubMed
Google Scholar
Cerruti Mainardi P: Синдром критического общения в этой взрослой жизни. Patologiagenica ad esordio tardivo. Под редакцией: Андрия Дж., Дагна Брикарелли Ф., Дель Порто Дж., Де Марчи М., Федерико А. Болонья: Мондузи; 1987: 113-128.
Google Scholar
Bruni L: La sindrome 5p- (sindrome del «cri du chat»).Malattie da aberrazioni cromosomiche. Под редакцией: Виньетти П., Ферранте Э. Торино: Edizioni Minerva Medica Italia; 1988, 89-94.
Google Scholar
Даллапиккола B: Синдром дель «кри дю чат». Difetti congeniti e sindromi malformative. Под редакцией: Мастрояково П., Даллапиккола Б., Андрия Г., Камера Г., Лунгаротти М.С. Милан: Макгроу Хилл Либри Италия; 1990, 254–255.
Google Scholar
Cerruti Mainardi P, Pastore G, Guala A: Синдром дель кри дю чат. Linee guida assistenziali nel bambino con sindrome malformativa. Под редакцией: Балестрацци П. Милано: CSH; 1994, 75-90.
Google Scholar
Cerruti Mainardi P, Perfumo C, Pastore G, Calì A, Guala A, Biroli E, Liverani ME, Egidi I, Zara F, Zerega G, Overhauser J, Pierluigi M, Dagna Bricarelli F: Cri du Chat Синдром. Ital J Pediatr. 2001, 27: 840-850. Http://www.ijp.it/articoli/2001/vol6-01/indice6_01.htm,
Google Scholar
Cerruti Mainardi P, Pastore G, Castronovo C, Godi M, Guala A, Tamiazzo S, Provera S, Pierluigi M, Dagna Bricarelli F: Естественная история синдрома Кри дю Чат. Отчет из Итальянского Регистра. Eur J Med Genet 2006 в печати.
Google Scholar
Rizzi M: Valutazione immologica in pazienti affetti dalla sindrome del cri du chat 5p-.Tesi di Laurea. Facoltà di Medicina e Chirurgia, Università degli Studi di Milano, Anno Accademico; 1997.
Google Scholar
Kjaer I, Niebuhr E: Исследования основания черепа у 23 пациентов с синдромом кри-дю-чат предполагают, что в это состояние вовлечено поле развития черепа. Am J Med Genet. 1999, 82: 6-14. 10.1002 / (SICI) 1096-8628 (199
) 82: 1 <6 :: AID-AJMG2> 3.0.CO; 2- #.
CAS
PubMed
Google Scholar
Marinescu RC, Cerruti Mainardi P, Collins MR, Kouahou M, Coucourde G, Pastore G, Eaton-Evans J, Overhauser J: Диаграммы роста для синдрома кри-дю-чат: международное совместное исследование. Am J Med Genet. 2000, 94: 153-162. 10.1002 / 1096-8628 (20000911) 94: 2 <153 :: AID-AJMG8> 3.0.CO; 2- #.
CAS
PubMed
Google Scholar
Коллинз М.С., Итон-Эванс Дж .: Исследование роста критического синдрома. Arch Dis Child. 2001, 85: 337-338.10.1136 / adc.85.4.337.
PubMed Central
CAS
PubMed
Google Scholar
Нибур Э. Антропометрия при синдроме Кри дю Шат. Clin Genet. 1979, 16: 82-95.
CAS
PubMed
Google Scholar
Breg WR, Steele MW, Miller OJ, Warburtonb D, Capoa A, Allerdice PW: Критический синдром у подростков и взрослых: клинические данные у 13 пожилых пациентов с частичной делецией короткого плеча хромосомы N. ° 5 (5р-).J Pediatr. 1970, 77: 782-791. 10.1016 / S0022-3476 (70) 80236-0.
CAS
PubMed
Google Scholar
Niebuhr E: Синдром кошачьего крика (5p-) у подростков и взрослых. J Ment Defic Res. 1971, 15: 277-291.
PubMed
Google Scholar
Van Buggenhout GJCM, Pijkels E, Holvoet M, Schaap C, Hamel BCJ, Fryns JP: Синдром Кри дю Чата: изменение фенотипа у пожилых пациентов.Am J Med Genet. 2000, 90: 203-215. 10.1002 / (SICI) 1096-8628 (20000131) 90: 3 <203 :: AID-AJMG5> 3.0.CO; 2-A.
CAS
PubMed
Google Scholar
Посмык Р., Панасюк Б., Яценко С.А., Станкевич П., Мидро А.Т .: Естественная история ребенка с синдромом моносомии 5р (синдром кошачьего крика / крик-дю-чат) в течение 18 лет наблюдения. вверх. Genet Couns. 2005, 16: 17-25.
CAS
PubMed
Google Scholar
Ховард РО: Окулярные аномалии при синдроме критичности. Am J Ophthalmol. 1972, 73: 949-954.
CAS
PubMed
Google Scholar
Kitsiou-Tzeli S, Dellagrammaticas HD, Papas CB, Ladas ID, Bartsocas CS: необычные глазные находки у младенца с синдромом кри-дю-чат. J Med Genet. 1983, 20: 304-307.
PubMed Central
CAS
PubMed
Google Scholar
Кобрински Л., Читаят Д., Захед Л., МакГрегор Д., Рочон Л., Браунштейн С., Векеманс М., Альберт Д.Л.: Трисомия 22 и фациоаурикуловертебральная последовательность (Голденхара). Am J Med Genet. 1993, 46: 68-71. 10.1002 / ajmg.1320460111.
CAS
PubMed
Google Scholar
Чунг Ю.Ф., Уоттс П., Литтл Э, Бек Л.: Синдромы Гольденхара и кри-дю-чат: синдром делеции непрерывного гена ?. J AAPOS. 2003, 7: 226-227.
PubMed
Google Scholar
McLellan MW, Golden WL, Wilson WG: синдромы Марфана и крид-чата у 18-месячного ребенка: доказательства взаимодействия фенотипов. Clin Genet. 1994, 46: 319-321.
Google Scholar
Stathopulu E, Mackie Ogilvie C, Flinter FA: Терминальная делеция хромосомы 5p у пациента с фенотипическими особенностями синдрома Лужана-Фринса. Am J Med Genet A. 2003, 119: 363-6. 10.1002 / ajmg.a.10268.
Google Scholar
Мартинес Дж. Э., Так-Мюллер К. М., Суперно Д., Вертелецкий В.: Фертильность и синдром критичности. Clin Genet. 1993, 43: 212-214.
CAS
PubMed
Google Scholar
Tamraz J, Rethoré MO, Lejeune J, Outin C, Goepel R, Stievenart JL, Iba-Zizen MT, Cabanis EA: Morphométrie Encéphalique en IRM dans la maladie du cri du chat. По поводу отдельных пациентов, avec revue de la littérature. Энн Жене. 1993, 36: 75-87.
CAS
PubMed
Google Scholar
De Michele G, Presta M, Di Salle F, Serra L, Mazzaccara A, Della Rocca G, Ambrosio G, Filla A: Гипоплазия червя мозжечка в случае синдрома кри-дю-чат. Acta Neurol (Неаполь). 1993, 15: 92-6.
CAS
Google Scholar
Balci S, Oguz KK: Синдром кри-дю-чат, связанный с арахноидальной кистой, вызывающий трехжелудочковую гидроцефалию. Clin Dysmorphol. 2001, 10: 289-290. 10.1097 / 00019605-200110000-00011.
CAS
PubMed
Google Scholar
Lejeune J, Rethoré MO, Peeters M, de Blois MC, Rabier D, Parvy P, Bardet J, Kamoun P: Maladie du cri du chat: acides aminés Plasmatiques et urinaires. Энн Жене. 1990, 33: 16-20.
CAS
PubMed
Google Scholar
Peeters MA, Rethoré MO, Aris L, Megarbane A, Cattaneo F, Lejeune J: метаболические аномалии в лимфоцитах кридочатного синдрома (5p-) и синтез новопуринов. Энн Жене. 1991, 34: 219-225.
CAS
PubMed
Google Scholar
Tsao CY, Wenger GD, Bartholomew DW: Синдром Кри-дю-чата и сложный кариотип у пациента с инфантильными спазмами, гипсаритмией, некетотической гиперглицинемией и гетеротопией. Am J Med Genet A. 2005, 134: 198-201. 10.1002 / ajmg.a.30592.
Google Scholar
Wilkins LE, Brown JA, Wolf B: Психомоторное развитие у 65 воспитываемых дома детей с синдромом кри-дю-чат. J Pediatr. 1980, 97: 401-405. 10.1016 / S0022-3476 (80) 80189-2.
CAS
PubMed
Google Scholar
Carlin ME: Улучшенный прогноз при синдроме кри-дю-чат (5p-). Материалы 8-го Конгресса Международной ассоциации научных исследований умственной отсталости. Под редакцией: Фрейзер В. 1990, Эдинбург: Блэквелл, 64-73.
Google Scholar
Корниш К.М., Пиграмма J: Характеристики развития и поведенческие характеристики критического синдрома.Arch Dis Child. 1996, 75: 448-450.
PubMed Central
CAS
PubMed
Google Scholar
Корниш К.М., Мунир Ф .: Рецептивные и выразительные речевые навыки у детей с синдромом кри-дю-чат. J Commun Disord. 1998, 31: 73-80. 10.1016 / S0021-9924 (97) 00052-X.
CAS
PubMed
Google Scholar
Корниш К.М., Брамбл Д., Мунир Ф., Пиграмма J: Когнитивные функции у детей с типичным критическим синдромом (5p-).Dev Med Child Neurol. 1999, 41 (4): 263-6. 10.1017 / S001216229
59.
CAS
PubMed
Google Scholar
Cerruti Mainardi P, Guala A, Pastore G, Pozzo G, Dagna Bricarelli F, Pierluigi M: Психомоторное развитие при критическом синдроме. Clin Genet. 2000, 57: 459-461. 10.1034 / j.1399-0004.2000.570612.x.
CAS
PubMed
Google Scholar
Франкенбург В.К., Доддс Дж.Б., Арчер П., Шапиро Х., Бресник Б.: Денвер II: обновленная стандартизация Денверского проверочного теста на развитие.Педиатрия. 1992, 89: 91-97.
CAS
PubMed
Google Scholar
Dykens EM, Clarke DJ: Корреляты дезадаптивного поведения у людей с синдромом 5p- (cri du chat). Dev Med Child Neurol. 1997, 39: 752-756.
CAS
PubMed
Google Scholar
Clarke DJ, Boer H: проблемное поведение, связанное с удалением синдромов Prader-Willi, Smith-Magenis и Cri du Chat.Am J Ment Retard. 1998, 103: 264-271. 10.1352 / 0895-8017 (1998) 103 <0264: PBAWDP> 2.0.CO; 2.
CAS
PubMed
Google Scholar
Коллинз М.С., Корниш К. Исследование распространенности стереотипов, членовредительства и агрессии у детей и молодых людей с синдромом Кри дю Ша. J Интеллект Disabil Res. 2002, 46: 133-140. 10.1046 / j.1365-2788.2002.00361.x.
PubMed
Google Scholar
Саримски К. Раннее игровое поведение у детей с синдромом 5p- (Кри-дю-Шат). J Интеллект Disabil Res. 2003, 47: 113-120. 10.1046 / j.1365-2788.2003.00448.x.
CAS
PubMed
Google Scholar
Gersh M, Goodart SA, Pasztor LM, Harris DJ, Weiss L, Overhauser J: данные об отдельной области, вызывающей кошачий крик у пациентов с делециями 5p. Am J Hum Genet. 1995, 56: 1404-1410.
PubMed Central
CAS
PubMed
Google Scholar
Church DM, Bengtsson U, Nielsen KV, Wasmuth JJ, Niebuhr E: Молекулярное определение делеций различных сегментов дистального 5p, которые приводят к различным фенотипическим особенностям. Am J Hum Genet. 1995, 56: 1162-1172.
PubMed Central
CAS
PubMed
Google Scholar
Church DM, Yang J, Bocian M, Shiang R, Wasmuth JJ: Физическая карта с высоким разрешением и транскриптом области Cri du Chat хромосомы 5p человека.Genome Res. 1997, 7: 787-801.
CAS
PubMed
Google Scholar
Cerruti Mainardi P, Perfumo C, Calì A, Coucourde G, Pastore G, Cavani S, Zara F, Overhauser J, Pierluigi M, Dagna Bricarelli F: Клиническая и молекулярная характеристика 80 пациентов с делецией 5p: генотип — фенотипическая корреляция. J Med Genet. 2001, 38: 151-158. 10.1136 / jmg.38.3.151.
Google Scholar
Simmons AD, Overhauser J, Lovett M: выделение кДНК из критической области Cri-du-chat путем прямого скрининга библиотеки кДНК, специфичной для 5 хромосомы. Genome Res. 1997, 7: 118-127.
CAS
PubMed
Google Scholar
Overhauser J, McMahon J, Oberlender S, Carlin ME, Niebuhr E, Wasmuth JJ, Lee-chen J: Родительское происхождение делеций хромосомы 5 при синдроме крид-чата. Am J Med Genet. 1990, 37: 83-86. 10.1002 / ajmg.1320370119.
CAS
PubMed
Google Scholar
Медина М., Маринеску Р.К., Оверхаузер Дж., Косик С.К .: Гемизигозность дельта-катенина (CTNND2) связана с тяжелой умственной отсталостью при синдроме кри-дю-чат. Геномика. 2000, 63: 157-164. 10.1006 / geno.1999.6090.
CAS
PubMed
Google Scholar
Overhauser J, Marinescu RC, Cheung M, Simmons A, Wixted D, Robin NH, Lovett M: Картирование генов в пределах уменьшенной критической критической области [аннотация].Am J Hum Genet. 1997, A136: 776.
Google Scholar
Hughes AE, McGibbon D, Woodward E, Dixey J, Doherty M: Локализация гена хондрокальциноза в хромосоме 5p. Hum Mol Genet. 1995, 4: 1225-1228.
CAS
PubMed
Google Scholar
Совместное исследование генетики астмы. Полногеномный поиск локусов восприимчивости к астме в этнически разнообразных популяциях.Nature Genet. 1997, 15: 389-392. 10.1038 / ng0497-389.
Google Scholar
Simmons AD, Pueschel AW, Mc Pherson JD, Overhauser J, Lovett M: Молекулярное клонирование и картирование человеческого семафорина F из интервала кандидатов в Кри-дю-Шат. Biochem Biophys Res Com. 1998, 242: 685-691. 10.1006 / bbrc.1997.8027.
CAS
PubMed
Google Scholar
Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, Liu X: Удаление нейрон-специфического белка дельта-катенина приводит к тяжелой когнитивной и синаптической дисфункции.Curr Biol. 2004, 14: 1657-1663. 10.1016 / j.cub.2004.08.065.
CAS
PubMed
Google Scholar
Zhang A, Zheng C, Hou M, Lindvall C, Li K, Erlandsson F, Björkholm M, Gruber A, Blennow E, Xu D: Удаление гена обратной транскриптазы теломеразы и гаплонедостаточность поддержания теломер в Cri. синдром дю чата. Am J Hum Genet. 2003, 72: 940-948. 10.1086 / 374565.
PubMed Central
CAS
PubMed
Google Scholar
Marinescu RC, Johnson EI, Dykens EM, Hodapp RM, Overhauser J: Нет связи между размером делеции и уровнем задержки развития при синдроме кри-дю-чат. Am J Med Genet. 1999, 86 (1): 66-70. 10.1002 / (SICI) 1096-8628 (199
) 86: 1 <71 :: AID-AJMG14> 3.0.CO; 2-Y.
CAS
PubMed
Google Scholar
Корниш К.М., Кросс Дж., Грин А., Уиллатт Л., Брэдш Дж. М.: Нейропсихолого-генетический профиль атипичного критического синдрома: значение для прогноза.J Med Genet. 1999, 36: 567-570.
PubMed Central
CAS
PubMed
Google Scholar
Niebuhr E: Цитологические наблюдения у 35 человек с 5p-кариотипом. Hum Genet. 1978, 42: 143-146. 10.1007 / BF00283634.
CAS
PubMed
Google Scholar
Baccichetti C: Del (5p) без фенотипа «cri du chat» [аннотация]. Hum Genet. 1982, 60: 389-10.1007 / BF00569228.
Google Scholar
Baccichetti C, Lenzini E, Artifoni L, Caufin D, Marangoni P: Терминальная делеция короткого плеча хромосомы 5. Clin Genet. 1988, 34: 219-223.
CAS
PubMed
Google Scholar
Overhauser J, Golbus MS, Schonberg SA, Wasmuth JJ: Молекулярный анализ несбалансированной делеции короткого плеча хромосомы 5, которая не дает фенотипа.Am J Hum Genet. 1986, 39: 1-10.
PubMed Central
CAS
PubMed
Google Scholar
Cerruti Mainardi P, Calì A, Guala A, Perfumo C, Liverani ME, Pastore G, Overhauser J, Zara F, Pierluigi M, Dagna Bricarelli F: фенотип-генотипическая корреляция у 7 пациентов с 5p / аутосомными транслокациями . Риск для носителей транслокаций с участием 5p [аннотация]. Am J Hum Genet. 2000, A753: 145.
Google Scholar
Perfumo C, Cerruti Mainardi P, Calì A, Coucourde G, Zara F, Cavani S, Overhauser J, Dagna Bricarelli F, Pierluigi M: первые три пациента с синдромом мозаичного кридука с двумя перестроенными клеточными линиями. J Med Genet. 2000, 37: 967-972. 10.1136 / jmg.37.12.967.
PubMed Central
CAS
PubMed
Google Scholar
Kitsiou S, Kolialexi A, Mavrou A: синдром Mosaic Cri du Chat у пациента с тремя клеточными линиями 5p.Prenat Diagn. 2004, 24: 578-579. 10.1002 / pd.906.
PubMed
Google Scholar
Леви Б., Данн Т.М., Керн Дж. Х., Хиршхорн К., Кардон Н. Б.: Определение фенотипа dup5q молекулярным цитогенетическим анализом у пациента с dup5q / del 5p (кри дю чат). Am J Med Genet. 2002, 108: 192-197. 10.1002 / ajmg.10261.
PubMed
Google Scholar
Росси Э., Де Грегори М., Патричелли М.Г., Прампаро Т., Арджентеро Л., Джильо С., Соста К., Форести Дж., Дзуффарди О: 8.Делеция 5 Mb в дистальной части 5p у мужчины с выявленной азооспермией. Am J Med Genet A. 2005, 133: 189-192. 10.1002 / ajmg.a.30519.
Google Scholar
Zhang X, Snijders A, Segraves R, Zhang X, Niebuhr A, Albertson D, Yang H, Gray J, Niebuhr E, Bolund L, Pinkel D: Картирование с высоким разрешением генотип-фенотипических отношений в кри синдром дю чата с использованием сравнительной геномной гибридизации массива. Am J Hum Genet. 2005, 76: 312-326.10.1086 / 427762.
PubMed Central
CAS
PubMed
Google Scholar
Wu Q, Niebuhr E, Yang H, Hansen L: Определение «критической области» для кошачьего крика синдрома Кри-дю-чат и анализ генов-кандидатов с помощью количественной ПЦР. Eur J Hum Genet. 2005, 13: 475-485. 10.1038 / sj.ejhg.5201345.
CAS
PubMed
Google Scholar
Harvard C, Malenfant P, Koochek M, Creighton S, Mickelson EC, Holden JJ, Lewis ME, Rajcan-Separovic E: вариант фенотипа Cri du Chat и расстройство аутистического спектра у субъекта с загадочными микроделециями de novo с участием 5п15.2 и 3p24.3-25 детектировали с использованием всего геномного массива CGH. Clin Genet. 2005, 67: 341-351. 10.1111 / j.1399-0004.2005.00406.x.
CAS
PubMed
Google Scholar
Marinescu RC, Johnson EI, Grady D, Chen XN, Overhauser J: FISH-анализ терминальных делеций у пациентов с диагнозом «синдром кри-дю-чат». Clin Genet. 1999, 56: 282-288. 10.1034 / j.1399-0004.1999.560405.x.
CAS
PubMed
Google Scholar
Granzow M, Popp S, Keller M, Holtgreve-Grez H, Brough M, Schoell B, Rauterberg-Ruland I, Hager HD, Tariverdian G, Jauch A: Мультиплексный анализ целостности теломер FISH выявляет несбалансированную криптическую транслокацию (5) t (3; 5) (q27; p15.3) в семье с тремя умственно отсталыми людьми. Hum Genet. 2000, 107: 51-57. 10.1007 / s0043
010.
CAS
PubMed
Google Scholar
Ensenauer R, Jalal S, Meyer R, Babovic-Vuksanovic D: Несбалансированная загадочная делеция 5p / дупликация 17p, идентифицированная субтеломерным FISH в семье с мальчиком с химеризмом и сбалансированным t (4; 5).Am J Med Genet A. 2004, 125: 86-91. 10.1002 / ajmg.a.20420.
Google Scholar
Кондо Т., Симокава О., Харада Н., Дои Т., Юн С., Годда Ю., Киношита Ф., Мацумото Н., Мориучи Н.: Генотип-фенотипическая корреляция 5p-синдрома: ловушка диагностики. J Hum Genet. 2005, 50: 26-29. 10.1007 / s10038-004-0213-9.
PubMed
Google Scholar
Бенн PA, Hsu LYF, Verma RS, Aloiso ML, Reich E, Wishnick M: Пренатальная диагностика минутной 5p-делеции: цитогенетическая проблема в обнаружении.Obstet Gynecol. 1987, 70: 449-452.
CAS
PubMed
Google Scholar
Smart RD, Retief AE, Overhauser J: Подтверждение сбалансированной хромосомной транслокации с использованием молекулярных методов. Prenat Diagn. 1989, 9: 505-513.
CAS
PubMed
Google Scholar
Bernstein R, Bocain ME, Cain MJ, Bengtsson U, Wasmuth JJ: Идентификация скрытой реципрокной транслокации t (5; 7) с помощью флуоресцентной гибридизации in situ.Am J Med Genet. 1993, 46: 77-82. 10.1002 / ajmg.1320460113.
CAS
PubMed
Google Scholar
Петтенати М.Дж., Хейворт Р., Кокс К., Рао П.Н.: Пренатальное выявление критического синдрома на некультивированных амниоцитах с использованием флуоресцентной гибридизации in situ (FISH). Clin Genet. 1994, 45: 17-20.
CAS
PubMed
Google Scholar
Chen CC, Lee CC, Chang TY, Town DD, Wang W: Пренатальная диагностика мозаичной дистальной делеции 5p и обзор литературы.Prenat Diagn. 2004, 24: 50-57. 10.1002 / pd.794.
PubMed
Google Scholar
Tullu MS, Muranjan MN, Sharma SV, Sahu DR, Swami SR, Deshmukh CT, Bharucha BA: Синдром Кри-дю-чат: клинический профиль и пренатальная диагностика. J Postgrad Med. 1998, 44: 101-104.
CAS
PubMed
Google Scholar
Аоки С., Хата Т., Хата К., Миядзаки К.: Антенатальные сонографические особенности синдрома кри-дю-чат.Ультразвуковой акушерский гинекол. 1999, 13: 216-219. 10.1046 / j.1469-0705.1999.13030216.x.
Google Scholar
Стефану Э.Г., Ханна Дж., Фоукс А., Крокер М., Фитчетт М.: Пренатальная диагностика синдрома кридуса (5p) в сочетании с изолированной умеренной двусторонней вентрикуломегалией. Prenat Diagn. 2002, 22: 64-66. 10.1002 / pd.243.
PubMed
Google Scholar
Сарно А.П., Ползин В.Дж., Калиш В.Б .: Сосудистое сплетение плода в сочетании с синдромом крид Чат (5p-).Am J Obstet Gynecol. 1993, 169: 1614-5.
PubMed
Google Scholar
Muller F, Aegerter P, Boue A: Перспективный скрининг хорионического гонадотропина в сыворотке крови матери на риск хромосомных аномалий плода и последующей внутриутробной и неонатальной смерти. Prenat Diagn. 1993, 13: 29-43.
CAS
PubMed
Google Scholar
Fankhauser L, Brundler AM, Dahoun S: Синдром кри-дю-чат, диагностированный с помощью амниоцентеза, выполненного из-за аномального анализа материнской сыворотки.Prenat Diagn. 1998, 18 (10): 1099-100. 10.1002 / (SICI) 1097-0223 (1998100) 18:10 <1099 :: AID-PD400> 3.0.CO; 2-H.
CAS
PubMed
Google Scholar
Вайс А., Шалев С., Вайнер Э, Шнеор Ю., Шалев Е. Пренатальная диагностика синдрома делеции 5p после аномально низкого уровня хорионического гонадотропина человека в материнской сыворотке. Prenat Diagn. 2003, 23: 572-574. 10.1002 / pd.645.
PubMed
Google Scholar
Vialard F, Robyr R, Hillion Y, Molina Gomes D, Selva J, Ville Y: синдром Денди-Уокера и агенезия мозолистого тела при делеции 5p. Prenat Diagn. 2005, 25: 311-313. 10.1002 / pd.1130.
CAS
PubMed
Google Scholar
Кушник Т., Рао К.В., Лэмб А.Н.: Семейный 5p-синдром. Clin Genet. 1984, 26: 472-476.
CAS
PubMed
Google Scholar
Bengtsson U, McMahon J, Quarrel Q, Rubenstein C, David K, Greenberg F, Wasmuth JJ: Фенотипически нормальные носители несбалансированной терминальной делеции 5p передают делеции потомству, у которого наблюдается задержка роста и развития [аннотация].Am J Hum Genet. 1990, A47: 208.
Google Scholar
Ямасита М., Таниока Ф, Танигучи К., Майсуки А., Ояма Т.: Анестезиологические соображения при синдроме крид-чата: отчет о трех случаях. Анестезиология. 1985, 63: 201-202.
CAS
PubMed
Google Scholar
Брислин Р.П., Стайер С.А., Шварц RE: Анестезиологические соображения для пациента с критическим синдромом.Педиатр Анаест. 1995, 5: 139-141.
CAS
PubMed
Google Scholar
Cerruti Mainardi P, Medolago LM, Pedrinazzi M: La Sindrome del Cri du Chat. 2002, Firenze: Grafiche Borri, S. Casciano V.P, http://www.criduchat.it/cdc/doc_vari/ABCLibrettoSCDC72/ABClibrettoSCDC72.pdf,
Google Scholar
Синдром кри-дю-чата | Причины и лечение
Синдром кри-дю-чата — хромосомная проблема, вызванная отсутствием части хромосомы 5.Синдром называется cri du chat (по-французски крик кошки), потому что пораженные дети часто издают пронзительный крик.
Не у всех младенцев с отсутствующим участком хромосомы 5 разовьется синдром критичности. Синдром кри-дю-чата может вызывать множество аномалий, особенно затрагивающих голову и лицо. Другие особенности могут включать трудности в обучении и медленный рост и развитие.
Специального лечения нет. Однако могут потребоваться физиотерапия, логопедия и языковая терапия, а также хирургическое лечение некоторых аномальных особенностей.Многие затронутые дети доживут до взрослой жизни. Однако такие дети могут умереть в течение первого года жизни.
Хромосомы и деление клетки
Хромосомы находятся в центре (ядре) клетки. Они несут генетическую информацию в виде генов. «Генетический» означает, что заболевание передается через семьи с помощью специальных кодов, называемых генами. Каждая клетка вашего тела содержит хромосомы, состоящие из множества генов.
В целом каждая клетка вашего тела содержит 46 хромосом, расположенных в 23 пары.Одна хромосома от каждой пары унаследована от вашей матери, а другая — от отца.
Одна из этих пар хромосом известна как половые хромосомы, потому что эта пара определяет наш пол. У женщин две половые хромосомы одного типа (XX). У мужчин две разные половые хромосомы (XY). Y-хромосома содержит гены, определяющие самцов. Итак, нормальной женщине 46, XX, а нормальному мужчине 46, XY. Остальные 22 пары хромосом пронумерованы в соответствии с размером: 1 — самая длинная пара, а 22 — самая короткая пара хромосом.
Однако яйцеклетки и сперматозоиды имеют только 23 хромосомы (по 1 хромосе из каждой пары). Это так, что, когда яйцеклетка и сперматозоид встречаются во время зачатия, у рожденного ребенка также есть 46 хромосом в каждой клетке.
Новые клетки организма производятся для роста и восстановления путем деления клеток с образованием двух «дочерних» клеток. Каждая хромосома может дублировать свою точную копию, так что каждая новая формирующаяся клетка имеет полный идентичный набор хромосом.
Редко бывает проблема с хромосомами ребенка.Возможные проблемы включают слишком много хромосом, слишком мало хромосом или повреждение одной или нескольких хромосом. Аномальные хромосомы могут означать, что ребенок не может выжить, что может вызвать выкидыш. Тем не менее, младенцы с некоторыми хромосомными аномалиями могут выжить, но у них будут возникать различные медицинские проблемы (так называемые синдромы).
Что вызывает синдром крик-дю-чата?
Синдром кри-дю-чата — хромосомная проблема, вызванная отсутствующим участком хромосомы 5. Отсутствующий участок хромосомы — это короткое (называемое «p») плечо хромосомы 5.Поэтому говорят, что синдром кридавата вызван делецией хромосомы 5р.
Считается, что в большинстве случаев это происходит в результате повреждения хромосомы во время развития яйцеклетки или сперматозоидов.
Каковы особенности синдрома кри-дю-чата?
Не у всех детей с отсутствующим коротким плечом хромосомы 5 разовьется синдром кридавы. Некоторые из них будут иметь только очень легкие аномальные черты или вообще не иметь аномальных черт.
Есть ряд признаков синдрома кри-дю-чата, в том числе:
- У ребенка высокий крик, который описывается как кошачий.Мякающий крик становится менее очевидным с возрастом.
- Проблемы с сосанием и кормлением — обычное явление в первый год жизни.
- Особенности головы ребенка могут включать маленькую головку (микроцефалия), маленькую челюсть (микрогнатия) и широко расставленные глаза.
- Аномальные черты лица также включают наклон вниз к глазам, низкие уши или уши неправильной формы и кожные бирки перед ухом. Может быть дополнительная складка кожи над внутренним уголком глаза (эпикантическая складка).
(Паола Черрути Майнарди, через Wikimedia Commons) - Аномальные особенности рук и ног включают частичное переплетение или соединение (слияние) пальцев рук или ног.На ладони может быть одна линия (складка) (обычно бывает две складки на коже).
- Пораженный новорожденный ребенок может быть маленьким и медленно расти. У пострадавшего ребенка могут быть трудности с обучением. Может наблюдаться медленное развитие моторики (например, задержка ходьбы), речи и языка.
- Другие признаки могут включать грыжу в паху и разделение мышц на животе. Также существует повышенный риск пороков сердца и аномалий головного мозга, почек или кишечника (кишечника).
Насколько распространен синдром критического разговора?
Синдром кри-дю-чата встречается очень редко. Он поражает примерно 1 из 30 000 новорожденных.
Как диагностируется синдром крид-чата?
Синдром Кри-дю-чат может быть диагностирован до рождения (пренатально) или после рождения (послеродовой).
Пренатальная диагностика
Диагноз до рождения можно поставить с помощью УЗИ или путем тестирования хромосом ребенка во время беременности. См. Отдельные буклеты «Амниоцентез, Взятие проб ворсинок хориона и ультразвуковое сканирование».
Послеродовая диагностика
Диагноз после рождения будет поставлен путем обследования, если у ребенка есть какие-либо признаки, указывающие на критический синдром. См. Отдельную брошюру под названием «Генетическое тестирование». Родители ребенка с критическим синдромом также должны пройти генетическое тестирование, чтобы выяснить, есть ли у одного из родителей изменение в хромосоме 5.
Потребуются дальнейшие тесты для оценки каких-либо признаков, связанных с критическим синдромом (например, череп Рентген и магнитно-резонансная томография (МРТ) для оценки любых аномалий черепа и лица или ультразвуковое сканирование сердца (эхокардиограмма) для оценки любых пороков сердца).
Как лечить синдром кри-дю-чат?
Не существует специального лечения синдрома крид-чата. Однако пострадавшим младенцам и детям может потребоваться серьезная физиотерапия, речевая и языковая терапия. Предоставление раннего специального образования и благоприятной домашней обстановки способствует развитию социальных и интеллектуальных способностей. Хирургическое лечение может потребоваться для исправления некоторых аномальных особенностей (например, грыжи) или любых других связанных функций (например, пороков сердца).
Каков прогноз при синдроме критичности?
Перспектива (прогноз) зависит от тяжести аномальных особенностей. Трудности в обучении и проблемы с речью и языком являются обычными. Однако большинство людей с синдромом крик-дю-чата доживают до взрослого возраста.
Около 1 из 10 детей, рожденных с синдромом кридука, серьезно страдают и умирают в течение первого года жизни. Умственная отсталость — обычное явление.
Серьезная инфекция легких (пневмония) чаще встречается из-за аномалий головы и лица.
Синдром кри-дю-чата | UF Health, Университет здравоохранения Флориды
Определение
Синдром кри-дю-чата — это группа симптомов, возникающих в результате отсутствия части хромосомы номер 5. Название синдрома основано на крике младенца, который является высоким и звучит как кошка.
Альтернативные названия
Синдром делеции хромосомы 5p; Синдром 5p минус; Синдром кошачьего крика
Причины
Синдром кри-дю-чата встречается редко. Это вызвано отсутствием участка хромосомы 5.
Считается, что в большинстве случаев это происходит во время развития яйцеклетки или сперматозоидов. В небольшом количестве случаев родитель передает ребенку другую перестроенную форму хромосомы.
Симптомы
Симптомы включают:
Обследования и анализы
Медицинский работник проведет медицинский осмотр. Это может показать:
Генетические тесты могут показать недостающую часть хромосомы 5. Рентгенография черепа может выявить любые проблемы с формой основания черепа.
Лечение
Специального лечения нет. Ваш врач порекомендует способы лечения или купирования симптомов.
Родители ребенка с этим синдромом должны пройти генетическое консультирование и тестирование, чтобы определить, есть ли у одного из родителей изменение хромосомы 5.
Группы поддержки
5P-Society — www.fivepminus.org
Перспективы (прогноз)
Умственная отсталость — распространенное явление. Половина детей с этим синдромом овладевает вербальными навыками, достаточными для общения.Кошачий крик со временем становится менее заметным.
Возможные осложнения
Осложнения зависят от степени умственной отсталости и физических проблем. Симптомы могут повлиять на способность человека заботиться о себе.
Когда обращаться к медицинскому работнику
Этот синдром чаще всего диагностируется при рождении. Ваш врач обсудит с вами симптомы вашего ребенка. После выписки из больницы важно продолжать регулярные посещения лечащего врача.
Генетическое консультирование и тестирование рекомендуется всем людям с семейным анамнезом этого синдрома.
Профилактика
Профилактика неизвестна. Пары с семейным анамнезом этого синдрома, желающие забеременеть, могут обратиться за генетической консультацией.
Ссылки
Bacino CA, Lee B. Cytogenetics. В: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, ред. Учебник педиатрии Нельсона . 20-е изд. Филадельфия, Пенсильвания: Эльзевьер; 2016: глава 81.
Мадан-Хетарпал С., Арнольд Г. Генетические нарушения и дисморфические состояния. В: Zitelli BJ, McIntire SC, Norwalk AJ, ред. Атлас детской физической диагностики Зителли и Дэвиса . 7-е изд. Филадельфия, Пенсильвания: Эльзевьер; 2018: глава 1.
Cri Du Chat — обзор
6.2.2.1 Когнитивные и адаптивные способности
Тяжелая умственная отсталость исторически считалась отличительной чертой CdLS (Ptacek et al., 1963). Однако в нескольких более поздних исследованиях сообщается, что показатели интеллекта варьируются от тяжелой до легкой и даже в пределах нормы (Ajmone et al., 2014; Клайн и др., 2007; Basile et al., 2007). В целом сейчас признано, что, хотя диапазон интеллекта, наблюдаемый при CdLS, широк, у большинства людей обнаруживаются интеллектуальные нарушения от умеренных до серьезных (Deardorff et al., 2016; Kline et al., 2018). Интересно, что существует определенная степень соответствия между умственной отсталостью и генетическими подтипами CdLS: например, люди с мутациями SMC1A обычно демонстрируют менее выраженную интеллектуальную отсталость, чем люди с мутациями NIPBL (Huisman et al., 2017).
Подробные исследования когнитивных способностей у людей с CdLS сообщают о нарушениях определенных типов когнитивных функций : например, сообщалось, что дефицит управляющих функций, таких как кратковременная зрительная память и умственная гибкость, более серьезен, чем можно ожидать, учитывая уровень умственной отсталости человека (Reid et al., 2017; Nelson et al., 2017). Эти же исследования также предполагают, что наблюдаемые профили нарушения управляющих функций могут коррелировать с профилями сложных поведенческих фенотипов, наблюдаемых при CdLS, такими как частота и тяжесть повторяющегося поведения и социальных нарушений (Reid et al., 2017; Нельсон и др., 2017). Недавнее исследование, в котором сравнивали подростков и взрослых с CdLS, которые были сопоставлены по возрасту, физической мобильности и способности восприятия речи с контрастной группой пациентов с синдромом Дауна, показало, что управляющие функции, такие как рабочая память, снижаются с возрастом у людей с CdLS, по сравнению лицам с синдромом Дауна (Reid et al., 2017). Это открытие предполагает, что определенные когнитивные функции могут резко снижаться с возрастом при CdLS, идея, подтвержденная предварительными данными, полученными на мышиных моделях дефицита Nipbl , которые показывают аномалии в различных популяциях клеток коры уже в 4-недельном возрасте (Santos, Calof и другие., в стадии подготовки и см. Рис. 6.6 ниже). Понимание того, почему снижение когнитивных способностей после подросткового возраста кажется резким у людей с CdLS, может дать новое понимание генетических факторов, которые способствуют старению мозга, а также предоставить информацию, полезную для лечения людей с синдромом.
У людей с CdLS значительно нарушена коммуникация: значительная часть детей с CdLS (более одной трети) не развивает речь, а у тех, кто действительно развивает только ограниченные вербальные коммуникативные навыки (Goodban, 1993; Sarimski, 1997; Kline et al., 2018; Бек, 1987). В случаях, когда у людей с CdLS действительно развивается речь, появление языка, как сообщается, значительно задерживается (Kline et al., 1993b). Несмотря на это, есть свидетельства того, что люди с CdLS обладают некоторыми восприимчивыми языковыми способностями, то есть способностью понимать язык (Ajmone et al., 2014; Hoddell et al., 2011). Коммуникативные способности у детей и подростков с CdLS отличаются от тех, которые наблюдаются при других синдромах развития, таких как синдром Кри-дю-Чат, даже когда испытуемые в сравнительных исследованиях соответствуют возрасту и когнитивным способностям (Hoddell et al., 2011). Это говорит о том, что некоторые коммуникативные дефициты при CdLS являются специфическими для синдрома, а не связаны с глобальными интеллектуальными и физическими нарушениями, вызванными синдромом, и делает понимание этиологии этих специфических дефицитов большим интересом для понимания того, как развиваются различия в общении.
Расстройство аутистического спектра ( ASD ) — подобные нарушения — включая плохие выразительные коммуникативные навыки, социальный дефицит, повторяющееся поведение и предпочтение жестких распорядков — распространены при CdLS (напр.г., Moss et al., 2008; Оливер и др., 2008; Moss et al., 2013a; Moss et al., 2012; Mulder et al., 2017; Наканиши и др., 2012). В недавнем метаанализе распространенность симптомов РАС у лиц с CdLS оценивалась в 43% (Richards et al., 2015), хотя некоторые исследования утверждали, что распространенность симптомов РАС у людей достигает 79%. с CdLS (Moss et al., 2013b). Симптоматика, подобная РАС, часто возникает при синдромах, при которых люди демонстрируют нарушение когнитивных способностей и восприимчивой речевой способности.Интересно, однако, что есть различия: исследование, сравнивающее людей с CdLS с людьми с синдромом Cri du Chat, в котором испытуемые были сопоставлены по уровню когнитивных способностей и восприимчивых языковых способностей, показало, что ASD-подобные характеристики более выражены при CdLS, чем можно было бы спрогнозировать с учетом наблюдаемой степени умственной отсталости (Moss et al., 2008). Кроме того, анализ синдромов показывает, что РАС проявляется с атипичным профилем в CdLS, так что люди с CdLS демонстрируют непропорционально большие трудности с навыками социальной коммуникации и испытывают более высокий уровень тревожности, чем люди с другими синдромами с симптомами, подобными РАС. общие (Cochran et al., 2015; Moss et al., 2013a; Оливер и др., 2011).
6.2.2.2 Поведенческие и эмоциональные особенности людей с CdLS
Повторяющееся поведение наблюдается более чем у половины детей и молодых людей с CdLS (Rojahn et al., 2013; Moss et al., 2009; Hyman et al. ., 2002; Градос и др., 2017). Наблюдалось несколько различных топографий повторяющегося поведения, включая раскачивание тела, причудливое положение тела и поворот предметов; также сообщалось о непропорциональной частоте ритуального поведения, такого как уборка и выстраивание предметов в ряд (Sarimski, 1997; Kline et al., 2018; Moss et al., 2009). Лица с CdLS также демонстрируют значительно больше компульсивного поведения, чем люди, соответствующие степени умственной отсталости, при этом упорядочивание и проверка компульсий считается обычным явлением (Oliver et al., 2008; Hyman et al., 2002).
Сложное поведение , , такое как самоповреждающее поведение , также часто встречается у детей с CdLS. Сообщается, что более половины детей с CdLS демонстрируют самоповреждающее поведение; Самоповреждающее поведение включает самоповреждение рук (например,ж., укусы), ударов и тряски головой (Berney et al., 1999; Oliver et al., 2009; Sloneem et al., 2009). Однако исследования случай-контроль показывают, что, хотя распространенность самоповреждающего поведения высокий, это может быть не первичный поведенческий фенотип, а скорее может быть вторичным по отношению к другим причинным механизмам, таким как болезненные состояния (например, гастроэзофагеальный рефлюкс), тревога и / или проблемы со сном, которые часто возникают у людей с CdLS ( Оливер и др., 2009; Оливер и др., 2017; Берни и др., 1999; Холл и др., 2008; Клайн и др., 2018; см. ниже).
Поведение, указывающее на тревогу часто проявляют люди с CdLS (Crawford et al., 2017). В частности, в ряде исследований было высказано предположение, что социальный профиль CdLS свидетельствует о социальной тревожности: в социальных ситуациях сообщалось о высоких показателях избирательного мутизма, крайней застенчивости, социального избегания, а также снижении вербализации и зрительного контакта (Moss et al. , 2016; Оливер и др., 2006; Ричардс и др., 2009). Повышенный когнитивный спрос также был описан как возможное событие, запускающее тревожное поведение при CdLS (Nelson et al., 2017). Наконец, при CdLS также сообщалось о поведении, указывающем на другие типы тревожных расстройств, включая тревогу разлуки, паническое расстройство и агорафобию (Crawford et al., 2017).
Появляются новые данные о связанных с возрастом — изменениях в поведении , эмоциях , и познании у людей с CdLS .В частности, сообщается, что пожилые люди испытывают большие трудности с исполнительными функциями, такими как вербальная рабочая память, и демонстрируют поведение, указывающее на тревогу и плохое настроение, чаще, чем у более молодых людей (Reid et al., 2017; Nelson et al., 2017; Moss и др., 2017). Хотя лонгитюдных исследований мало, результаты исследований с использованием анализа возрастных групп показывают, что подростковый возраст / ранняя взрослая жизнь, вероятно, является критическим периодом для неблагоприятных изменений в познании, поведении и эмоциях при CdLS.Это привело к осознанию того, что у людей с CdLS могут быть значительные возрастные изменения нервной функции, которые только недавно начали исследоваться (Kline et al., 2018). Понимание раннего начала возрастных изменений нейронных и когнитивных функций при CdLS должно оказаться полезным не только для достижения лучшего качества ухода и качества жизни для людей с CdLS и их лиц, осуществляющих уход, но также для получения нового понимания факторов, которые способствуют возникновение возрастных неврологических и психических заболеваний, широко распространенных среди населения.
Синдром кри-дю-чат: основы практики, патофизиология, эпидемиология
Автор
Митилеш Кумар Лал, MD, MBBS, MRCP, FRCPCH, MRCPCH (Великобритания) Консультант неонатолог, клинический директор по неонатальным службам, младший медицинский директор (повторная валидация), Университетская больница Джеймса Кука, Доверительный фонд NHS South Tees Foundation; Британский председатель, экзамен по теории MRCPCH, старший клинический экзаменатор MRCPCH, Королевский колледж педиатрии и здоровья детей (Великобритания)
Митилеш Кумар Лал, доктор медицины, MBBS, MRCP, FRCPCH, MRCPCH (Великобритания) является членом следующих медицинских обществ: Педиатрическое общество, Общество педиатрических исследований, Британская ассоциация перинатальной медицины, Британская медицинская ассоциация, Неонатальное общество, Непальская медицинская ассоциация, Королевский колледж педиатрии и здоровья детей, Королевский колледж врачей
Раскрытие информации: раскрывать нечего.
Специальная редакционная коллегия
Мэри Л. Виндл, PharmD Адъюнкт-профессор, Фармацевтический колледж Медицинского центра Университета Небраски; Главный редактор Medscape Drug Reference
Раскрытие информации: нечего раскрывать.
Главный редактор
Мария Декарт, доктор медицины Профессор, кафедра генетики человека и кафедра педиатрии, Университет Алабамы, Медицинская школа Бирмингема
Мария Декарт, доктор медицины, является членом следующих медицинских обществ: Американская академия педиатрии, Американский медицинский колледж Генетика и геномика, Американская медицинская ассоциация, Американское общество генетики человека, Общество наследственных нарушений обмена веществ, Международное общество скелетной дисплазии, Юго-восточная региональная генетическая группа
Раскрытие: нечего раскрывать.
Дополнительные участники
Гарольд Чен, доктор медицины, магистр медицины, FAAP, FACMG Профессор, кафедра педиатрии, Медицинский центр Университета штата Луизиана
Гарольд Чен, доктор медицины, магистр медицины, FAAP, FACMG является членом следующих медицинских обществ: Американская академия педиатрии, Американский колледж медицинской генетики и геномики, Американская медицинская ассоциация, Американское общество генетики человека
Раскрытие информации: нечего раскрывать.